





Abstract
A hallmark of craniofacial development is the differentiation of multiple cell lineages in close proximity to one another. The mouse skull bones and overlying dermis are derived from the cranial mesenchyme (CM). Cell fate selection of the embryonic cranial bone and dermis in the CM requires Wnt/β-catenin signaling, and loss of β-catenin leads to an ectopic chondrogenic cell fate switch. The mechanism by which Wnt/β-catenin activity suppresses the cartilage fate is unclear. Upon conditional deletion of β-catenin in the CM, several key determinants of the cartilage differentiation program, including Sox9, become differentially expressed. Many of these differentially expressed genes are known targets of the Polycomb Repressive Complex 2 (PRC2). Thus, we hypothesized that PRC2 is required for Wnt/β-catenin-mediated repression of chondrogenesis in the embryonic CM. We find that β-catenin can physically interact with PRC2 components in the CM in vivo. However, upon genetic deletion of Enhancer of Zeste homolog 2 (EZH2), the catalytic component of PRC2, chondrogenesis remains repressed and the bone and dermis cell fate is preserved in the CM. Furthermore, loss of β-catenin does not alter either the H3K27me3 enrichment levels genome-wide or on cartilage differentiation determinants, including Sox9. Our results indicate that EZH2 is not required to repress chondrogenesis in the CM downstream of Wnt/β-catenin signaling.
Embryonic craniofacial development involves the formation of complex structures from two progenitor stem cell pools: the cranial neural crest (CNC) and cephalic paraxial mesoderm (PM) (Jiang et al. 2002; Yoshida et al. 2008). The CNC gives rise to the more anterior tissues of the head and the PM gives rise to the posterior tissues. Both the CNC and PM contribute to the mesenchymal stem cells surrounding the brain called the CM. The cranial bones and the overlying dermis are derived from the CM (Yoshida et al. 2008; Tran et al. 2010). For proper patterning and development of these tissues, specific signaling pathways are required to regulate cell fate selection and tissue morphogenesis (Fan et al. 2016). Disruption of these pathways can lead to craniofacial malformations such as craniosynostosis and focal dermal hypoplasia (Wilkie 1997; Wang et al. 2007; Hsu et al. 2010). Thus, understanding the genetic mechanisms directing skull and dermal cell fate selection is critical to elucidating the etiology of such malformations.
The Wnt/β-catenin pathway is instructive for cranial bone and dermal fibroblast cell fate selection in the developing embryo (Atit et al. 2006; Ohtola et al. 2008; Tran et al. 2010; Goodnough et al. 2012; Fan et al. 2016). Conditional loss of ectodermal Wnt-ligand secretion or mesenchymal β-catenin leads to a loss of cranial bones and dermis. Instead, an ectopic formation of cartilage, marked by the upregulation of a key chondrocyte marker gene Sox9, is observed (Tran et al. 2010; Goodnough et al. 2012, 2014; Budnick et al. 2016). In intramembranous bone, loss-of-function mutations in other known signaling pathways important in craniofacial development, such as those driven by Fibroblast Growth Factors and Bone Morphogenetic Proteins, do not result in ectopic chondrogenesis (O’Rourke and Tam 2002; Fan et al. 2016). In craniofacial development, Wnt/β-catenin signaling seems to have a unique role in the repression of chondrogenesis in the CM.
β-catenin is a central transducer of the canonical Wnt signaling pathway, where it acts as a transcriptional coactivator of context-specific target genes to regulate cell fate selection in many cell types during development (Bhanot et al. 1996; Korinek et al. 1998; Liu et al. 1999; Haegele et al. 2003; Verani et al. 2007). While β-catenin is typically known as a transcriptional activator, a stabilized or post-translationally methylated form of β-catenin has been shown to function as a transcriptional repressor in vitro (Delmas et al. 2007; Hoffmeyer et al. 2017). However, the mechanism by which Wnt/β-catenin signaling in the CM prevents chondrogenesis, while ensuring proper cranial bone and dermal fibroblast cell fate selection in vivo, is unknown.
Recent in vitro studies have suggested epigenetic histone modifications, by the PRC2 specifically, as a possible mechanism by which Wnt/β-catenin signaling represses chondrogenesis. PRC2 is a multi-protein complex that is required for the repressive histone modification H3K27me3 (Jiang et al. 2002; Lund and Van Lohuizen 2004; Peng et al. 2009). In multiple cell types and organisms, numerous connections between the Wnt/β-catenin pathway and PRC2 have been demonstrated. First, like Wnt/β-catenin signaling, PRC2 is required for the regulation of cell fate selection (Lee et al. 2006; Sparmann and van Lohuizen 2006; Asp et al. 2011; Margueron and Reinberg 2011). Second, Sox9 and other chondrocyte differentiation determinants are known targets of PRC2 by H3K27me3 enrichment in multiple cell types ranging from mouse embryonic stem cells (ESCs) to chick limb bud micromass cultures (Peng et al. 2009; K. I. Kim et al. 2013; Kumar and Lassar 2014; Tien et al. 2015). Third, PRC2 regulates components of the Wnt/β-catenin pathway and vice versa (Wang et al. 2010; Zemke et al. 2015; Mirzamohammadi et al. 2016; Yi et al. 2016). Fourth, β-catenin can physically interact with PRC2 components (Shi et al. 2007; Li et al. 2009; Jung et al. 2013; Hoffmeyer et al. 2017). Fifth, β-catenin and PRC2 can cooperate with one another to enhance either Wnt signaling or PRC2 activity (Shi et al. 2007; Jung et al. 2013; Kumar and Lassar 2014; Hoffmeyer et al. 2017). It is important to note that these studies were all performed in cell culture models with one or more overexpressed proteins. Follow-up studies in vivo are therefore required. Understanding how Wnt/β-catenin signaling intersects with PRC2 to direct cell fate selection in vivo will provide new insights into the genetic mechanisms governing cranial bone and dermal development.
Here, we test the hypothesis that repression of chondrogenesis in the CM by Wnt/β-catenin signaling requires PRC2-mediated epigenetic repression. In a conditional β-catenin loss-of-function model, among the genes dysregulated in both mutant CM and mutant dorsal mesenchyme, we found an overrepresentation of known targets of the PRC2 pathway. Conditional deletion of Ezh2 in the CM does not phenocopy the ectopic cartilage in the β-catenin mutants, nor do H3K27me3 levels change upon complete loss of β-catenin in the CM. Our results suggest that the repression of chondrogenesis in the CM is not reliant on PRC2, indicating that repressive mechanisms besides PRC2 are likely involved. We propose that the “off” state of chondrogenic genes is not actively maintained by PRC2 and that β-catenin represses chondrogenesis by regulating an unidentified inhibitory pathway.
Materials and Methods
Mice and genotyping
The following strains were used in this study: Engrailed1Cre (En1Cre) (Kimmel et al. 2000), Rosa26 Reporter (R26R) (Soriano 1999), β-catenin null (β-catenin∆) (Brault et al. 2001), conditional β-catenin floxed (β-cateninfl) (Haegel et al. 1995), Twist2Cre(Dermo1Cre) (Yu et al. 2003), and conditional Ezh2 floxed (Ezh2fl) (Shen et al. 2008). Mice were maintained in mixed genetic backgrounds. For timed matings, En1Cre;β-catenin+/∆ males were crossed with R26R/R26R;β-cateninfl/fl females, and Dermo1Cre;Ezh2fl/+ males were crossed with Ezh2fl/fl females. Vaginal plugs were checked every morning and assigned as embryonic (E) 0.5. For each experiment, a minimum of three mutants with litter-matched controls were studied unless otherwise noted. Animals of both sexes were randomly assigned to all the studies. The Case Western Reserve University (CWRU) Institutional Animal Care and Use Committee approved all animal procedures in accordance with AVMA guidelines (Protocol 2013-0156, approved November 21, 2014, Animal Welfare Assurance No. A3145-01).
CM isolation
At E13.5, the CM was isolated by manual dissection. An incision was made around the circumference of the CM and the tissue covering the brain was manually dissociated. The CM samples were a mixed cell population comprised of the CNC- and PM-derived CM, which is En1Cre-positive, and also contained the overlying ectoderm, which is negative for En1Cre (CM+ectoderm). Each embryo yielded ∼500,000 CM cells for the controls and 250,000–500,000 CM cells for the mutants. Individual embryos were kept separate and considered as single biological replicates. The wild-type samples isolated for co-immunoprecipitation were dissociated by incubating the tissue in 0.25% Trypsin-EDTA (Thermo Fisher Scientific 25200056) at 37° for 5–7 min, and the CM was selectively enriched from the ectoderm using an Invitrogen FlowComp Flexi Kit (Invitrogen 11060D) and a PDGFRα antibody (5–10 µg/2.5 million cells) (R&D Systems AF1062) (Goodnough et al. 2016) according to the manufacturer’s guidelines.
RT-qPCR
The CM+ectoderm was manually dissected from E13.5 embryos (described above). RNA was isolated as previously described (Hamburg-Shields et al. 2015). Relative mRNA expression levels were quantified using 5 ng of cDNA on a StepOne Plus Real-Time PCR System (Life Technologies) and the ΔΔCT method. Commercially available TaqMan probes (Life Technologies) specific to each gene were used: Ezh2 (Mm00468464_m1), Suz12 (Mm01304152_m1), Eed (Mm00469660_m1), Sox9 (Mm00448840_m1), Axin2 (Mm00443610_m1) Twist2 (Mm00492147_m1), Keratin14 (mm00516876_m1), and Pdgfrα (Mm00440701_m1). CT values obtained for specific genes were normalized to those of β-actin (Invitrogen 4352663).
Immunofluorescence
Heads of E13.5 embryos were fixed in 4% paraformaldehyde for 30 min at 4° and cryopreserved as previously described (Atit et al. 2006). Rabbit polyclonal antibodies against H3K27me3 (1:1000; Cell Signaling 9733), LEF1 (1:100; Cell Signaling 2286), SP7/OSX (1:1000; Abcam ab94744), and SOX9 (1:1000; Millipore ab5535) were used for indirect immunofluorescence assays. Appropriate species-specific Alexafluor 594 secondary antibodies were used (1:500; Invitrogen). Images were captured using an Olympus BX60 microscope and an Olympus DP70 digital camera using DC controller software. Confocal images were captured on a Leica TCS SP8 (Leica Biosystems) using Application Suite X software (Leica Biosystems). Images were processed using ImageJ/Fiji (Schindelin et al. 2012; Schneider et al. 2012) and Adobe Photoshop software. Images were prepared for cell counting in ImageJ/Fiji by subtracting the background and thresholding the signal across all replicates. The percent of the cells that were H3K27me3-positive compared to DAPI was determined using the “analyze particles” feature in ImageJ/Fiji. Counting was performed on the supraorbital CM directly above the eye.
Co-immunoprecipitation
E13.5 CM was collected by manual dissection and the CM was enriched from the ectoderm (described above). At least 2 million cells from multiple embryos were incubated with 1 ml lysis buffer (50 mM Tris pH 7.5, 250 mM NaCl, 2 mM EGTA, and 1% Triton X-100 in H2O) and disrupted with a 23-gauge needle and syringe. The precipitating antibody was added to the lysate and incubated overnight at 4°. Immunoprecipitation was performed by incubating 100 µl of Dynabeads Protein G beads (Invitrogen 10003D) with the lysate and antibody for 1 hr at 4°. Beads were then washed four times with 1 ml wash buffer (40 mM HEPES, 300 mM NaCl, 10% Glycerol, and 0.2% NP40 in H2O). The sample was collected in NuPage LDS Sample Buffer (Thermo Fisher Scientific NP0007) supplemented with β-mercaptoethanol, and was heated at 90° for 5 min. Rabbit polyclonal antibodies against nonphospho-β-catenin, 3.43 µg (Cell Signaling D13A1 #8814); EZH2, 10 µg (Diagenode C15410039); and IgG, 6 µg (Abcam ab46540) were used for immunoprecipitation. Protein species were separated by SDS-PAGE using Mini-PROTEAN TGC gels (Bio-Rad #456-1084). Western blots were performed with primary antibodies against β-catenin (1:1000; Millipore 06-734) or EZH2 (1:500, Cell Signaling #5246). Clean-Blot IP Detection Reagent (HRP) (1:250; Thermo Fisher Scientific 21232) was used as secondary antibody.
Protein isolation and immunoblotting
E13.5 CM+ectoderm was collected by manual dissection. Protein was isolated using RIPA buffer (Cell Signaling 9806). Protein species were separated by SDS-PAGE using Mini-PROTEAN TGC gels (Bio-Rad #456-1084). Western blots were performed with polyclonal rabbit primary antibodies against H3K27me3 (1:1000, Cell Signaling 9733), EZH2 (1:500, Cell Signaling #5246), and SUZ12 (1:1000, Cell Signaling 3737). Species-specific HRP-conjugated secondary antibodies were used at a 1:10,000 dilution. Immunoblots were probed with a rabbit anti-β-tubulin antibody (1:400, Santa Cruz 9104) as a loading control. Signals were detected using an Amersham ECL Western Blotting Analysis System (GE Healthcare RPN2109), and imaged using an Odyssey FC Imaging System (Li-Cor). Relative protein levels were quantitated using ImageJ/Fiji software (Schindelin et al. 2012; Schneider et al. 2012).
RNA sequencing
E13.5 CM+ectoderm was collected by manual dissection (described above). Total RNA was isolated from individual embryos as previously described (Hamburg-Shields et al. 2015). Libraries were prepared in the CWRU Genomics sequencing core using the Illumina TruSeq Stranded Total RNA kit-with Ribo Zero Gold. Paired-end sequencing was performed on an Illumina HiSequation 2500 v2 Rapid Run flow cell. The resulting 100 bp reads were aligned to the mouse mm9 assembly using TopHat (Trapnell et al. 2009; Kim and Salzberg 2011; Langmead and Salzberg 2012; D. Kim et al. 2013). Genomic assembly was completed using Cufflinks v1.3 (Trapnell et al. 2010, 2013, Roberts et al. 2011a,b). mm9_reFlat was used to annotate the data with a maximum intron length of 20,000 bp and genomic bias correction. Cufflinks FPKMs < 0.3 were floored to 0.3. Differential gene expression was determined with CuffDiff using the default settings plus genomic bias correction. Gene ontology analysis examining all differentially expressed genes was performed using Genomic Regions Enrichment of Annotations Tool (GREAT) by associating reads to the single nearest gene located within 5 kb (McLean et al. 2010).
ChIP-seq
E13.5 CM+ectoderm was manually dissected from three En1Cre;β-cateninfl/+ and four En1Cre;β-cateninfl/∆ embryos, pooled, and H3K27me3 immunoprecipitation and sequencing was performed by Active Motif (www.activemotif.com) (deposited in GEO, GSE96872). Next, 14 µg chromatin was immunoprecipitated with 4 µg rabbit anti-H3K27me3 (Millipore #07-449). Sequencing was performed on an Illumina NextSequation 500 producing 75-nucleotide, single-end reads. Drosophila DNA was “spiked in.” The ratio of aligned Drosophila reads in the mutant vs. control samples (calculated to be 1.3) was used to normalize the number of reads in the mouse samples by downsampling the larger sample (mutant, in this case). Sequences were aligned and analyzed twice independently. The analysis was first performed using a custom pipeline consisting of Bowtie2 for genome alignment to the mouse mm9 genome and Macs 1.4 at default settings for peak calling (Zhang et al. 2008; Langmead and Salzberg 2012). To generate the windowed heat map from this analysis, the genome was divided into 40 windows of equal size 5 kb up- and downstream of each H3K27me3 peak genome-wide or on peaks located within 1 kb of known promoters. The median peak signal in each window was then converted to a z-score and mapped using Java TreeView (Saldanha 2004). The analysis was performed a second time using the NGS 2.8 pipeline (Strand NGS Manual, Version 2.8, Build 230243, Strand Life Sciences, Bangalore, India) and aligning to the mm10 genome. Peaks were called using Macs 1.4 at default settings. Association of peaks with specific genes was performed using PAVIS (Huang et al. 2013). Specific H3K27me3 peaks were visualized using the Integrated Genome Viewer (IGV) (Robinson et al. 2011; Thorvaldsdóttir et al. 2013). Ngs.plot was used to generate the average fold enrichment of H3K27me3 overview across the gene bodies (Shen et al. 2014).
Cell culture
The CM+ectoderm was manually isolated and dissociated by incubating the tissue in 0.25% Trypsin-EDTA (Thermo Fisher Scientific 25200056) at 37° for 5–7 min, and then plated in DMEM with 10% fetal bovine serum. Fibroblasts were allowed to adhere to the plate for 1–2 hr, after which the media was removed and fresh media was added. Chemical inhibition was performed at no later than passage 3. Next, 10% Wnt3a-conditioned media and the chemical inhibitor, UNC1999 (Sigma SML0778) or GSK126 (Cayman Medical CAS1346574-57-9), were added simultaneously. The cells were incubated for the indicated amount of time. Following incubation, the cells were trypsinized and processed for protein or mRNA analysis.
Statistics
Graphs and statistical analyses were generated using Prism 6 (GraphPad Software). Data are presented as mean ± SEM in all graphs unless otherwise stated. All pairwise sample comparisons were performed using a Mann–Whitney test. The P-values for statistical tests in all figures are represented as * P < 0.05 and ** P < 0.01.
Data availability
Strains are publicly available at the Jackson Laboratory. Sequencing data are available at GEO with the accession number GSE96872.
Results
Genes dysregulated upon loss of β-catenin are enriched for the PRC2-associated H3K27me3 histone mark
In an effort to determine a functional link between β-catenin and PRC2 in vivo, we conditionally deleted β-catenin in the CM using Engrailed1Cre (En1Cre), manually dissected the CM along with the ectoderm (CM+ectoderm), and collected all the CNC- and PM-derived mesenchyme surrounding the brain (Figure 1A) (Kimmel et al. 2000; Tran et al. 2010). En1Cre is expressed in both the CNC- and PM-derived CM. In order to analyze in vivo tissues with minimal manipulation, the ectoderm was isolated with the CM. We then profiled the whole transcriptome on three litter-matched E13.5 En1Cre/+;R26R/+;β-cateninfl/+ controls and four En1Cre/+;R26R/+;β-cateninfl/Δ mutants using the RNA-seq approach (GSE96872). The analysis of the data revealed 521 genes that were differentially expressed by at least 1.4-fold in the two experimental groups (P < 0.05). Of the 521 differentially expressed genes, 322 were downregulated and 199 were upregulated in the mutants relative to the controls. Validating the approach, changes in expression of known Wnt/β-catenin targets were observed despite the presence of ectodermal cells, in which canonical Wnt signaling is known to be active (Supplemental Material, Figure S1A in File S1 [all Supplemental legends are in File S2] (Budnick et al. 2016). To ascertain the function of all the 521 differentially expressed genes, we performed a gene ontology analysis using GREAT, which queries multiple ontology databases (McLean et al. 2010). As a comparison, we also analyzed RNA-seq data from E13.5 En1Cre/+;R26R/+; β-cateninfl/Δ dorsal dermal mesenchyme (GSE75944) (Budnick et al. 2016). The top five ontologies of the differentially expressed genes in both the mutant CM+ectoderm and mutant dorsal dermal fibroblasts included the Wnt signaling pathway, along with Cadherin signaling, Integrin signaling, and ECM-receptor interactions (Figure S1, B and C in File S1) (Thomas et al. 2003). In the Molecular Signatures Database (MsigDB) Perturbations ontology, we also found that both data sets were highly enriched for genes regulated by PRC2 (Figure 1B and Figure S2A in File S1) (Subramanian et al. 2005). Interestingly, enrichment for targets of PRC2 can be found in both the up- and downregulated genes. However, this enrichment is unique only to the genes differentially expressed in our β-catenin mutants. GREAT analysis on all genes expressed in the CM+ectoderm (FPKM ≥ 1) did not result in enrichment for targets of PRC2 in the MSigDB Perturbations ontology (Figure S2B in File S1). Thus, the differential expression of PRC2 targets in the β-catenin mutant CM+ectoderm and dorsal mesenchyme reveals a potential functional link between the two pathways.
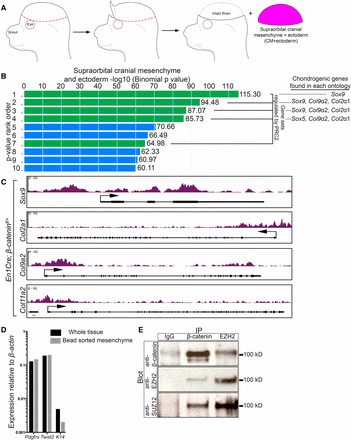
β-catenin regulates known PRC2 targets and can physically interact with PRC2. (A) Schematic demonstrating the isolation of CM+ectoderm by manual dissection. The isolated tissue is comprised of the supraorbital CM and overlying ectoderm. (B) Gene ontology analysis of differentially expressed genes in E13.5 En1Cre/+;R26R/+;β-cateninfl/+ control (n = 5) and En1Cre/+;R26R/+;β-cateninfl/∆ mutant CM+ectoderm (n = 4). Overlapping gene sets from MsigDB Perturbations ontology ranked by binomial P-value, which accounts for varying sizes of gene regulatory domains, from GREAT. Green bars represent gene sets associated with PRC2. The ranked list is as follows: (1) Genes with high-CpG-density promoters bearing H3K4me2 and H3K27me3 in the brain (MsigDB M1941). (2) Genes with H3K27me3 on their promoters in human embryonic stem cells identified by ChIP on chip (MsigDB M10371). (3) Genes identified as targets of SUZ12 in human embryonic stem cells by ChIP on chip (MsigDB M2291). (4) Genes identified as targets of EED in human embryonic stem cells by ChIP on chip (MsigDB M2736). (5) Genes coordinately upregulated in a compendium of adult tissue stem cells (M1999). (6) Genes upregulated in uterus upon knockout of BMP2 in the uterus (MsigDB M2324). (7) Genes downregulated in mouse embryonic stem cells upon deletion of SUZ12 (MsigDB M2291). (8) Genes upregulated in human lung fibroblasts (IMR90) after knockdown of RB1 by RNAi (MsigDB M2129). (9) Genes downregulated in breast cancer (MsigDB M13072). (10) Genes downregulated in immortalized nontransformed mammary epithelium (HMLE) after knockdown by RNAi or expression of a dominant negative form of CDH1 (MsigDB M11790). (C) Integrated Genome Viewer representation of H3K27me3 ChIP-sequencing on E13.5 En1Cre/+;R26R/+;β-cateninfl/+ control CM+ectoderm of Sox9 (chr11:112,641,524–112,651,071), Col2a1 (chr15:97,804,033–97,837,155), Col9a2 (chr4:120,710,171–120,729,930), and Col11a2 (chr17:34,174,382–34,205,187). (D) RT-qPCR of enriched CM after bead purification with an antibody specific to PDGFRα. A reduction in K14 expression levels demonstrates purification of the mesenchyme. (E) Co-immunoprecipitation of β-catenin with components of PRC2. CM+ectoderm was isolated by manual dissection and the mesenchyme was purified using a PDGFRα antibody. IgG was used as a negative immunoprecipitation control. ChIP, chromatin immunoprecipitation; chr, chromosome; CM, cranial mesenchyme; MsigDB, Molecular Signatures Database; PRC2, Polycomb Repressive Complex 2; RNAi, RNA interference; RT-qPCR, reverse transcriptase-quantitative polymerase chain reaction.
Chondrocyte fate genes are enriched for H3K27me3 in the embryonic CM
To establish a role for PRC2 in the repression of chondrogenesis in the CM in vivo, we queried for H3K27me3 enrichment in the loci of individual chondrocyte marker genes. We manually dissected the CM+ectoderm in E13.5 En1Cre/+;R26R/+;β-cateninfl/+ controls and performed ChIP using an antibody against H3K27me3 followed by massive parallel DNA sequencing (ChIP-seq). This assay allowed us to unbiasedly and comprehensively map the genome-wide distribution of the H3K27me3 modification (Active Motif Technology) (GSE96872). In the CM+ectoderm of E13.5 controls, the transcriptional start sites of multiple cartilage markers, such as Sox9, Col2a1, Col9a2, and Col11a2 (Figure 1C), were enriched for H3K27me3, indicating that they are targets of PRC2 in the CM.
Endogenous β-catenin and EZH2 may physically interact in the CM
Given the emerging connections made between the Wnt/β-catenin pathway and PRC2 in various systems in vitro (summarized in Table S1 in File S1), we set out to test the hypothesis that β-catenin and PRC2 components physically interact at native protein levels in the mouse CM extracts. We manually dissected the CM, made a cell suspension, and used a PDGFRα antibody bound to magnetic beads to enrich for the CM population (Goodnough et al. 2016). We found comparable levels of mRNA for mesenchyme progenitor markers Pdgfrα and Twist2, and diminished ectoderm marker Keratin 14 (K14) in the purified sample, confirming enrichment for CM (Figure 1D). We then prepared cell extracts from sorted CM and used them in a co-immunoprecipitation assay for β-catenin and EZH2. EZH2 is the methlytransferase component of PRC2, and is required for the H3K27me3 modification (Margueron and Reinberg 2011). In line with our hypothesis, β-catenin successfully co-immunoprecipitated with the EZH2 antibody. In addition, we also observed reciprocal co-immunoprecipitation of EZH2 and another major PRC2 component, SUZ12, by the β-catenin antibody (Figure 1E). These results suggest that PRC2 components and β-catenin may physically interact at wild-type expression levels in the CM. Thus, these data provide a potential molecular link between Wnt/β-catenin signaling and PRC2 in the mouse embryo.
β-catenin is not required for PRC2 component expression or bulk H3K27me3 levels
To determine if β-catenin is required for the formation of the PRC2 complex itself, we first examined the expression of the main PRC2 components: Ezh2, Suz12, and Eed. Based on FPKM values from our RNA-seq data set, we found no significant changes in the PRC2 component mRNA levels (Figure 2A). To validate this result, we manually dissected E13.5 En1Cre/+;R26R/+;β-cateninfl/+ control and En1Cre/+;R26R/+;β-cateninfl/Δ mutant CM+ectoderm (Figure 1A), and determined the mRNA levels of Ezh2, Suz12, and Eed by RT-qPCR. Similar to the RNA-seq data set, the relative mRNA levels of the individual PRC2 components were comparable in the control and mutant samples (Figure 2B). In comparison, the expected changes in mRNA levels were observed in known β-catenin responsive genes Axin2 and Sox9 (Figure 2, A and B) (Jho et al. 2002; Goodnough et al. 2012). Evaluation of the total H3K27me3 and EZH2 protein levels using western blot assays also revealed comparable protein levels between control and β-catenin mutant CM+ectoderm (Figure 2, C and D). To obtain spatial information and account for levels in the ectoderm between our control and mutants, we performed indirect immunofluorescence for H3K27me3 in the E13.5 En1Cre/+;R26R/+;β-cateninfl/+ control and En1Cre/+;R26R/+;β-cateninfl/Δ mutants (Figure 2, E–G). While we consider the CM to include the entire CM surrounding the brain (Figure 1A), we focused our indirect immunofluorescence analysis on the region directly above the eye (supraorbital CM) (Figure 2E) due to easily identified histological landmarks such as the eye and brain ventricles. Considering that knockout of β-catenin results in ectopic chondrogenesis throughout the CM, we expect the supraorbital CM to be representative of the entire CM. In the supraorbital CM, both the control and β-catenin mutants are positive for H3K27me3, demonstrating that PRC2 is still active without β-catenin. Furthermore, H3K27me3 can still be found in the expanded SOX9 domain in the β-catenin mutants. We concluded that β-catenin is not required cell autonomously in the CM to regulate the relative mRNA levels of major PRC2 components, the EZH2 protein levels, and bulk H3K27me3 levels. However, these results leave open the possibility that it may be required to recruit PRC2 to site-specific loci on the genome.
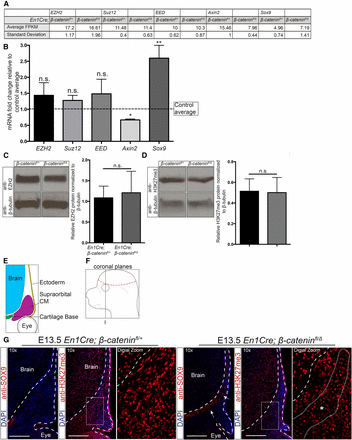
β-catenin activity is not required for the expression of PRC2 components and bulk H3K27me3 levels. (A) FPKM values obtained from RNA sequencing assays in CM+ectoderm from En1Cre/+;R26R/+;β-cateninfl/∆ mutant and En1Cre/+;R26R/+;β-cateninfl/+ controls. (B) RT-qPCR analysis of the CM+ectoderm in En1Cre/+;R26R/+;β-cateninfl/+ (n = 7) and En1Cre/+;R26R/+;β-cateninfl/∆ mutants (n = 9). The data are represented as fold change in mutants over controls. The dotted line represents the β-catenin controls. Axin2 and Sox9 are known targets regulated by β-catenin. (C and D) Western blot analysis (n = 5) of EZH2 and H3K27me3 in CM+ectoderm from En1Cre/+;R26R/+;β-cateninfl/∆ mutants and En1Cre/+;R26R/+;β-cateninfl/+ controls. Band intensities were quantified using ImageJ. (E and F) Schematics representing the supraorbital CM in coronal sections near the frontal bone primordia. (G) Indirect immunofluorescence of SOX9, H3K27me3, and DAPI in the supraorbital mesenchyme (n = 2 controls; 3 mutants). Images were taken near the frontal bone primordia (plane I). Dashed lines indicate the brain and ectoderm boundaries. (*) indicates region of ectopic cartilage. Bar, 200 µm. CM, cranial mesenchyme; DAPI, 4’,6-diamidino-2-phenylindole; FPKM, fragments per kilobase of transcript per million mapped reads; n.s., not significant; RT-qPCR, reverse transcriptase-quantitative polymerase chain reaction.
Loss of Ezh2 does not lead to ectopic cell type fate selection or chondrogenesis in the CM
We next determined if PRC2 is required for the repression of chondrogenesis in the CM in vivo. In order to remove PRC2 function in the CM, we conditionally deleted Ezh2 using a floxed allele (Shen et al. 2008). Surprisingly, conditional deletion of Ezh2 at E10.5 using En1Cre did not lead to the expected loss of H3K27me3 in the CM by indirect immunofluorescence (Figure S3 in File S1). We then conditionally deleted Ezh2 in the CM using Dermo1Cre, which is expressed in the CM by E10.0 (Yu et al. 2003; Goodnough et al. 2012). Loss of Ezh2 was sufficient to lead to an upregulation of Cdkn2a, a known target of PRC2 (Figure 3A) (Shen et al. 2008; Lui et al. 2016). We also found depletion of H3K27me3 in the supraorbital CM (Figure 3C) by indirect immunofluorescence between the Dermo1Cre; Ezh2fl/fl mutants to Dermo1Cre; Ezh2fl/+ controls (Figure 3, B and D). H3K27me3 signal was maintained in both the ectoderm and the brain, where Dermo1Cre is not expressed. After confirming the absence of PRC2 activity, we then examined the protein level of cell fate markers for bone, dermis, and cartilage progenitors by indirect immunofluorescence. Conditional deletion of Ezh2 in the supraorbital CM did not lead to changes in the location and size of the dermal domain as indicated by LEF1, and the bone domain as indicated by Osterix (OSX) (Figure 3, E and F). Consistently, we did not observe ectopic expression beyond the cartilage base of the key cartilage differentiation determinant SOX9 (Figure 2G). Based on these data, Ezh2 has little effect on the patterning of the tissue domains and minimal effect on the protein expression levels by immunofluorescence.
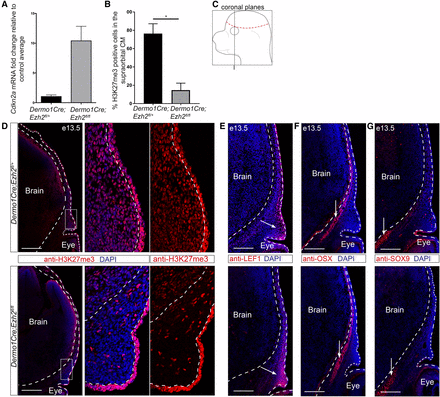
Knockdown of Ezh2 in the cranial mesenchyme does not lead to changes in cell fate selection. (A) Known downstream target Cdkn2a expression relative to controls in Dermo1Cre;Ezh2fl/fl CM+ectoderm (n = 2). (B) Percent H3K27me3 positive cells in the supraorbital CM. (C) Schematic representing the coronal sections near the frontal bone primordia. (D–G) Indirect immunofluorescence in Dermo1Cre;Ezh2fl/fl supraorbital CM. (D) Qualitative loss of the PRC2 repressive mark H3K27me3. (E–G) Domains of similar size and location were observed for the osteoblast (OSX), dermal fibroblast (LEF1), and chondrocyte (SOX9) markers between controls and mutants. Dashed lines indicate the brain and ectoderm boundaries. (D–F) Arrows mark the tissue domains. Bar, 200 µm. CM, cranial mesenchyme; DAPI, 4’,6-diamidino-2-phenylindole; PRC2, Polycomb Repressive Complex 2. * P < 0.05.
To further test the H3K27me3-dependent role of PRC2 in the repression of chondrogenesis, we chemically inhibited EZH2 function in primary E13.5 CM+ectoderm cells in vitro (Figure 4, A and E). Incubation with small molecule methyltransferase inhibitor GSK126, which is specific for EZH2, or with UNC1999, which inhibits EZH2 and EZH1, led to a considerable reduction in bulk H3K27me3 protein levels (Figure 4, B and F). Upon treatment with GSK126 or UNC1999, the Sox9 and Col2a1 mRNA levels were not significantly increased (Figure 4, C, D, and G). Taken together, these data indicate that EZH2 and H3K27me3 are dispensable for regulating the mRNA level of chondrocyte differentiation markers in the CM+ectoderm.
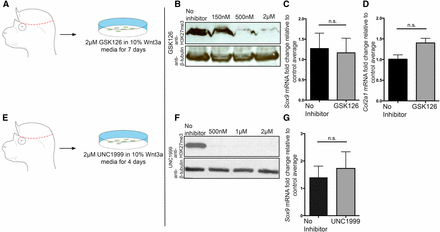
Chemical inhibition of EZH2 methyltransferase does not lead to an upregulation of early chondrocyte markers in CM+ectoderm. (A and E) Schematic demonstrating the isolation of primary CM+ectoderm fibroblasts. GSK126 is specific to EZH2, and UNC1999 is specific to both EZH2 and EZH1. GSK126 and UNC1999 inhibit EZH2’s methyltransferase activity. (B and F) Western blots demonstrating reduction/loss of H3K27me3 level following incubation with GSK126 ((IC50 = 75–100 nm) or UNC1999 (IC50 < 10 nM for EZH2 and 45 nM for EZH1). (C, D, and G) qPCR analysis of the expression of Sox9 and Col2a1 following inhibition of EZH2. GSK126: n = 5 mutants and 6 controls for Sox9, and n = 3 mutants and controls for Col2a1. UNC1999: n = 7 mutants and 9 controls. CM, cranial mesenchyme; n.s., not significant; qPCR, quantitative polymerase chain reaction. * P < 0.05; ** P < 0.01.
Loss of β-catenin does not significantly alter H3K27me3 enrichment genome-wide
Next, we tested to what extent β-catenin is required for the recruitment of PRC2 to the genome in a site-specific manner. We performed ChIP-seq assays, as described in Figure 1, to map the genome-wide distribution of H3K27me3 in the CM in vivo between En1Cre/+;R26R/+;β-cateninfl/+ controls and En1Cre/+;R26R/+;β-cateninfl/Δ mutants (GSE96872). Sequencing of the CM+ectoderm revealed, by two independent analyses, 14,337 peaks in the control and 10,752 peaks in the mutant, thus 25% fewer peaks in the mutant. Surprisingly, genome-wide comparisons between individual mutant and control H3K27me3 peaks revealed modest differences in enrichment fold between the two samples (Figure 5A). The differences in peak numbers between β-catenin controls and mutants were associated with changes in smaller H3K27me3 peaks (Figure 5, A’, A’’’, B’, and B’’’). Furthermore, any gains and losses of strength of H3K27me3 peaks were not associated with gene expression changes (Figure 5B). In addition, on all genes bound by H3K27me3, the signal intensity of the peaks was also comparable between the mutant and control across the gene body (Figure 5C). Next, we examined changes in H3K27me3 peak signal across the gene body of the differentially expressed genes identified in our RNA-seq data. In both the up- and downregulated genes, the H3K27me3 enrichment was comparable between β-catenin controls and mutants (Figure 5D).
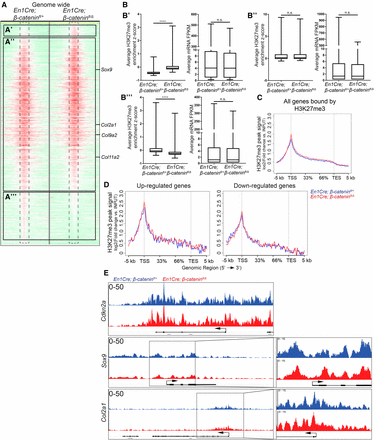
Loss of β-catenin does not significantly alter H3K27me3 enrichment genome-wide or on cartilage differentiation determinants. (A) Treeview representation of H3K27me3 peak strength genome-wide in the CM+ectoderm between E13.5 En1Cre/+;R26R/+;β-cateninfl/+ controls and En1Cre/+;R26R/+;β-cateninfl/∆ mutants. H3K27me3 ChIP-sequencing signal strength was mapped 5 kb up- and downstream from each peak. A change from high intensity to low intensity identifies a peak is lost. (B) Intersection of changes in H3K27me3 enrichment from (A), with En1Cre/+;R26R/+;β-cateninfl/∆ mutant and En1Cre/+;R26R/+;β-cateninfl/+ control RNA-seq (B’–B’’’) corresponding with (A’–A’’’), respectively. (C and D) Intersection of En1Cre/+;R26R/+;β-cateninfl/+ control and En1Cre/+;R26R/+;β-cateninfl/∆ mutant ChIP-sequencing and RNA-seq. H3K27me3 ChIP-sequencing signal strength was measured across all genes bound by H3K27me3 or genes identified to be differentially expressed in β-catenin mutant CM+ectoderm. The x-axis demarcates the percent distance across a gene between the TSS and the TES. (E) IGV representation of H3K27me3 signal peaks between En1Cre/+;R26R/+;β-cateninfl/+ control (n = 1) and En1Cre/+;R26R/+;β-cateninfl/∆ mutant (n = 1) CM+ectoderm. Cdkn2a is a known target of PRC2. Sox9 and Col2a1 are chondrocyte marker genes. ChIP, chromatin immunoprecipitation; CM, cranial mesenchyme; FPKM, fragments per kilobase of transcript per million mapped reads; n.s., not significant; RNA-seq, RNA-sequencing; TES, transcription end site; TSS, transcription start site.
From our ChIP-seq data set, we observed variations of the H3K27me3 enrichment throughout the genome ranging from large peaks blanketing an entire gene body to smaller peaks located just on the promoter. To further investigate the connection between H3K27me3 peak enrichment strength and gene expression, we divided the H3K27me3 peaks into three categories based on the level of enrichment: strong (>20-fold enrichment), medium (10–20-fold enrichment), and weak (≤ fivefold enrichment) (Figure S4 in File S1). Representative enrichment for strong, medium, and weak peaks can be found on the HoxA cluster, Sept9, and Lmtk3, respectively (Figure S4A in File S1). The genomic location of each class of peak using GREAT revealed that the large majority of strong and medium peaks were within 5 kb of the transcription start site (TSS), and the weak peaks had a more even distribution spanning out 500 kb from the TSS (Figure S4B in File S1). Between controls and β-catenin mutants, the number of strong and medium peaks was comparable, with most of the variation found in the weak peaks (Figure S4C in File S1). To further characterize each class of peak, we performed gene ontology analysis on the control H3K27me3 ChIP-seq data set. Gene ontology analysis revealed distinct functions for the strong peaks such as DNA-binding/transcription regulation and conserved homeobox sites, and the medium and weak peaks shared functions such as Wnt signaling and ion transport (Figure S5 in File S1). Furthermore, comparisons between each class of peak found near a TSS (±5 kb) and the genes identified in our RNA-seq data set revealed that 70% of strong peaks, 53% of medium peaks, and 47% of weak peaks were associated with transcriptional repression (<1 FPKM) (Figure S6A in File S1). These results indicate that the level of H3K27me3 enrichment may be predictive of its transcriptional repressive function. When we intersected genes bound by each class of H3K27me3 peak and differentially expressed genes, we found that each class of peak had similar enrichment between the down- and upregulated genes, indicating that H3K27me3 enrichment does not predict transcriptional repression by β-catenin (Figure S6B in File S1).
H3K27me3 enrichment is not depleted on ectopically-expressed chondrocytic gene determinants in β-catenin mutants
To determine if the loss of β-catenin resulted in depletion of H3K27me3 on chondrocyte differentiation determinants, we examined the enrichment of H3K27me3 on Sox9 and its downstream target Col2a1, which have higher mRNA levels in the β-catenin mutant CM (Figure 2) (Goodnough et al. 2012). Cdkn2a, HoxA, and T/Brachyury are known targets of PRC2, contain strong H3K27me3 peaks, and serve as controls. Upon deletion of β-catenin, we did not observe a change in H3K27me3 enrichment on known PRC2 target genes (Figure 5E and Figure S7, A and B in File S1). More importantly, H3K27me3 enrichment did not change on Sox9, Col2a1, Col9a2, or Col11a2 (Figure 5E and Figure S7, H and I in File S1). Furthermore, H3K27me3 enrichment was similar between β-catenin controls and mutants on the TSS of critical bone and dermal marker genes, such as Runx2, Twist1, Twist2, Axin2, and Lef1 (Figure S7, C–G in File S1). Mcm6 serves as a negative control and lacks H3K27me3 enrichment in either the control or mutant (Figure S7J in File S1). It is worth noting that Cdkn2a, HoxA, and T loci had strong H3K27me3 enrichment peaks, while Sox9 and Col2a1 loci had medium enrichment peaks in both controls and β-catenin mutants (Figure 5E and Figure S7, A and B in File S1). Our results showed that H3K27me3 enrichment is not depleted from the TSS of chondrocyte differentiation determinants in β-catenin mutants and remains enriched in actively transcribed genes.
Discussion
Based on in vivo evidence that β-catenin is required to repress chondrogenesis in the CM, and on emerging in vitro evidence connecting β-catenin and PRC2 in other processes, we tested the hypothesis that repression of chondrogenesis by Wnt/β-catenin signaling requires epigenetic repression by PRC2 in vivo. Consistent with the findings from previous studies, our results demonstrate that an in vivo loss of β-catenin in the CM and dorsal mesenchyme leads to the activation of chondrogenic marker genes such as Sox9, Col2a1, and Col11a2, as well as other known PRC2 target genes. Further, we find that β-catenin can physically interact with PRC2 components at native protein levels in CM-enriched protein extracts. In contrast to findings from in vitro studies, we observe that β-catenin is not required for the expression of major PRC2 components in vivo and that PRC2 is dispensable for the repression of chondrogenic marker genes in CM cells. Conditional deletion of β-catenin in the CM does not alter H3K27me3 enrichment around differentially expressed genes nor genome-wide in vivo. Our data in genetic mutants in vivo are consistent with a model whereby EZH2 and H3K27me3 are not required in the CM to guide cell fate selection. Interrogating mixed cell populations is unlikely to account for our major finding, given that our CM-restricted deletion of β-catenin did not lead to changes in H3K27me3 profiles, and Ezh2 mutants in vivo did not show changes in cell fate selection in the supraorbital CM.
Considering that loss of β-catenin at E10.5 leads to ectopic chondrogenesis, but loss of Ezh2 at E10.5 did not phenocopy the β-catenin mutant, the function of the physical interaction between β-catenin and PRC2 remains unclear. A recent study in human colon cancer cells demonstrated that EZH2 alone, independent of H3K27me3, was sufficient to repress transcription (O’Geen et al. 2017). While we did not observe genome-wide changes in H3K27me3 enrichment upon loss of β-catenin, it is possible that β-catenin is required to recruit EZH2 itself to the genome. Alternatively, EZH2 was recently shown to bind to β-catenin in mouse ESCs and trimethylate lysine 49 (K49me3) on the β-catenin protein itself (β-catMe3) (Hoffmeyer et al. 2017). The β-catMe3 protein could then function as a transcriptional repressor at defined loci in ESCs to govern neuronal vs. mesoderm fate. However, loss of Ezh2 in the CM did not lead to alteration in cell fate selection, indicating that K49me3 modification of β-catenin does not play a role in cell fate selection in the CM. Future studies examining DNA binding by EZH2 and β-catenin could provide a biological function for the physical interaction between β-catenin and EZH2.
The lack of cell fate changes in the supraorbital CM of Ezh2 mutants could indicate that the role of EZH2, and by extension PRC2, is dependent on the developmental stage and cell type. Most studies linking PRC2 and cell fate selection were performed in ESCs in vitro. Differences in the role of EZH2 between in vivo CM and in vitro ESCs may indicate that the cell fate selection role of PRC2 is unique to ES cells or linked to specific cell types. In addition, previous studies deleting Ezh2 at similar developmental stages in the mouse embryo found varying craniofacial phenotypes and defects (Schwarz et al. 2014; Dudakovic et al. 2015). Deletion of Ezh2 in the premigratory cranial neural crest cells with Wnt1Cre by E8.5 resulted in severe reduction of facial and skull bones, and embryonic lethality (Schwarz et al. 2014). Conditional deletion of Ezh2 at E9.5 in posterior CM with Prx1Cre resulted predominantly in postnatal craniosynostosis. We did not find gross changes in embryonic craniofacial morphology upon deletion of Ezh2 in the CM at E10.0 with Dermo1Cre (data not shown). These results suggest that the role of PRC2 in embryonic development may be cell type- and developmental stage-specific. Future studies in vivo are required to tease out the timing and dynamics of developmental gene regulation by PRC2.
There are recent data from several groups refining the role of PRC2 and H3K27me3 enrichment. According to the histone code hypothesis, H3K27me3 is often found on transcriptionally repressed genes and is widely considered to be a sign of transcriptional repression (Boyer et al. 2006; Lee et al. 2006; Roh et al. 2006; Barski et al. 2007; Heintzman et al. 2007, 2009). In early postmigratory mouse neural crest cells, H3K27me3 was shown to mark bivalent domains accessible by the activating mark H3K4me3, indicating transcriptional poising rather than repression (Minoux et al. 2017). Recently, the histone code model has been refined to show that H3K27me3 enrichment is not just predictive of transcriptional repression in mouse ESCs, but also indicative of a past transcriptional repressive state (Riising et al. 2014; Comet et al. 2016). Furthermore, in human colon cancer cells, ectopic deposition of H3K27me3 with an EZH2-dCas9 fusion construct was not sufficient for transcriptional repression (O’Geen et al. 2017). In mouse rib chondrocytes, an intersection of RNA-seq data with H3K27me3 ChIP-seq data also suggested that H3K27me3 enrichment on TSS was not sufficient for transcriptional repression. When compared to genes dysregulated upon knockout of a major PRC2 component, EED, only 11% of the genes dysregulated were enriched for H3K27me3. Thus, the biological role of the remaining 89% of H3K27me3 peaks is unclear (Mirzamohammadi et al. 2016). Our data are entirely consistent with these recent findings. Intersecting our in vivo RNA-seq and ChIP-seq studies revealed legitimate H3K27me3 peaks in genes that did not correlate with transcription levels. We found that both expressed and repressed genes in control CM+ectoderm can be enriched for H3K27me3, demonstrating that H3K27me3 is not sufficient to indicate repression. The H3K27me3 marks remain at Sox9 and other cartilage marker genes in β-catenin mutants, suggesting that these marks may be carried over and reflective of a past transcriptional off state.
If PRC2 is not the principal repressor of chondrogenesis in CM, the question remains as to what factor exerts this function. We propose three other models that will require further testing. The first model calls for other epigenetic-related mechanisms such as direct covalent modifications of DNA (DNA methylation), or other histone modification-related mechanisms, such as G9a-associated K9me3 repression. A study in chick limb bud micromass cultures showed that the addition of exogenous Wnt3a led to an increase in DNA methylation by DNMT3a on the Sox9 promoter (Kumar and Lassar 2014). However, in our hands, the addition of DNMT inhibitors did not alter Sox9 and Col2a1 mRNA levels in primary CM+ectoderm cells cultured in vitro (data not shown). Further studies in vivo will be required to investigate this model. The second model postulates that β-catenin activates yet-to-be-identified signaling pathways or transcription factors that would be directly involved in repression. For example, Twist1 is positively regulated by Wnt/β-catenin signaling, and conditional deletion of Twist1 partially phenocopies the ectopic chondrogenesis found in the En1Cre/+;R26R/+;β-cateninfl/Δ mutants (Komori et al. 1997; Goodnough et al. 2012). The retinoic acid (RA) signaling pathway can interact with Wnt/β-catenin signaling, and it can promote chondrocyte development and function in vitro (Yasuhara et al. 2010; Uchibe et al. 2017). RA signaling pathway components are robustly expressed in the control CM. Their interaction with Wnt/β-catenin signaling and the role in the CM remains to be tested. A third model is that β-catenin does not directly control the transcription of cartilage determinants and marker genes, but may control the expression or activity of factors involved in the post-transcriptional modification of cell fate determination and chondrocyte differentiation genes.
Overall, our data suggested a model whereby the repression of the chondrogenic fate by Wnt/β-catenin signaling does not rely on EZH2 and H3K27me3, but implies other yet-to-be-identified transcriptional or post-transcriptional mechanisms.
Acknowledgments
We thank previous and current members of the Atit laboratory for excellent discussion and advice, and the Case Microscopy and Genomics Cores Services. This work was supported by the following grants: National Institutes of Health (NIH) T32 AR-007505 (J.F.), NIH National Institute of Dental and Craniofacial Research R01 DE-01870 (R.P.A.), NIH R01 DE-01870, NIH R01 CA-160356 (P.C.S.), NIH R01 CA-193677 (P.C.S.), NIH National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) R01 AR-46249 (V.L.), NIH-NIAMS R01 AR-68308 (V.L.), and the Case Western Reserve University ENGAGE (M.D.) and SOURCE Programs (M.D.).
Author contributions: J.F. and R.P.A. conceived experiments; J.F., M.D., and R.P.A. carried out the experiments; J.F., A.S., C.-F.L., and R.P.A. analyzed the data; G.D. performed genotyping and westerns; J.F., A.S., and C.-F.L. generated figures; J.F., V.L., P.C.S., and R.P.A. interpreted the data and wrote the article. The authors declare that they have no competing interests.
Footnotes
Supplemental material is available online at www.g3journal.org/lookup/suppl/doi:10.1534/g3.117.300311/-/DC1.
Communicating editor: M. Boutros

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}