





Abstract
All genomes contain repeated sequences that are known as transposable elements (TEs). Among these are endogenous retroviruses (ERVs), which are sequences similar to retroviruses and are transmitted across generations from parent to progeny. These sequences are controlled in genomes through epigenetic mechanisms. At the center of the epigenetic control of TEs are small interfering RNAs of the piRNA class, which trigger heterochromatinization of TE sequences. The tirant ERV of Drosophila simulans displays intra-specific variability in copy numbers, insertion sites, and transcription levels, providing us with a well-suited model to study the dynamic relationship between a TE family and the host genome through epigenetic mechanisms. We show that tirant transcript amounts and piRNA amounts are positively correlated in ovaries in normal conditions, unlike what was previously described following divergent crosses. In addition, we describe tirant insertion polymorphism in the genomes of three D. simulans wild-type strains, which reveals a limited number of insertions that may be associated with gene transcript level changes through heterochromatin spreading and have phenotypic impacts. Taken together, our results participate in the understanding of the equilibrium between the host genome and its TEs.
All genomes contain repeated sequences that are known as transposable elements (TEs). These are sequences that can move and multiply along the chromosome arms, generating most of the time deleterious mutations. A few decades ago, TEs were only acknowledged as parasitic “junk DNA”; however, we now have evidence that host-TE relationships may range from parasitism to mutualism, as do all symbiotic interactions (Biémont and Vieira 2006; Jangam et al. 2017). TEs may be of various structures (Wicker et al. 2007), among which are endogenous retroviruses (ERVs). ERVs are sequences similar to retroviruses and are transmitted across generations from parent to progeny. Due to the potential harmful activity of TEs, mechanisms that allow to control them have been favored by natural selection, and consist mainly of epigenetic processes (Slotkin and Martienssen 2007; Siomi et al. 2011). In Drosophila, knowledge of such processes is increasingly accumulating. At the center of the epigenetic control of TEs are small interfering RNAs of the piRNA class, which trigger heterochromatinization of TE sequences through trimethylation of histone 3 lysine 9 (H3K9me3) (Sienski et al. 2012; Le Thomas et al. 2013).
TE impacts on genomes have varied nature and extents, from deleterious to adaptive (reviewed in Cordaux and Batzer 2009; Casacuberta and González 2013; Fablet et al. 2017). For instance, TE insertions may cause diseases (Hancks and Kazazian 2016), whereas in some cases, TEs may be recruited by the host genome and perform endogenous functions. The most famous example is the syncytin gene in mammals, which is an ancient TE gene and is now essential for placenta formation (Mi et al. 2000). TEs may also be involved in adaptation. As an example, the Accord TE inserted into the 5′ end of the Cyp6g1 gene of Drosophila melanogaster is found in some strains and provides them with increased resistance to insecticides (Daborn et al. 2002).
Despite their potential harmful impacts, TEs may reach high proportions of genome sequences (Biémont and Vieira 2006; Tenaillon et al. 2010; Elliott and Gregory 2015). Therefore, it is of fundamental interest to precisely understand TE dynamics within genomes, especially at the transcriptional level, which is the preliminary stage before mobilization. In this study, we propose to use the tirant element of Drosophila simulans to tackle this important issue.
Tirant belongs to the ERV class of retrotransposons (Terzian et al. 2001). It is made of three open reading frames (ORFs), namely gag, pol, and env, which provide all proteins necessary to fulfill the retroviral cycle, and is bordered by two long terminal repeats (LTRs), which include regulatory signals. Two subfamilies can be distinguished based on their sequence similarity and the structure of a minisatellite located in the 5′ untranslated region (UTR) (Fablet et al. 2006). Subfamily C is responsible for tirant activity while subfamily S is heterochromatic, low-copy-number, and silent (Fablet et al. 2006, 2009). Tirant was first discovered in D. melanogaster (Garrell and Modolell 1990) and found to occur in related species (Fablet et al. 2007). Its number of copies and insertion sites, together with expression profiles vary across D. simulans wild-type strains (Vieira et al. 1999; Fablet et al. 2006; Akkouche et al. 2012), providing a powerful system to investigate the impacts of ERVs on the host genome, particularly regarding neighboring gene expression and epigenetic signatures.
The analyses of RNA-seq and small RNA-seq data indicate that tirant transcript amounts and piRNA amounts are positively correlated in ovaries in normal conditions, while they were previously found to be negatively correlated in some divergent crosses leading to tirant deregulation. These results allow us to propose that the shape of the correlation between the amounts of TE transcripts and the corresponding piRNAs are indicative of genome stability. In addition, we describe tirant insertion polymorphism in the genomes of three D. simulans wild-type strains, which reveals a limited number of insertions that may be associated with gene transcript level changes through heterochromatin spreading and have phenotypic impacts. Taken together, our results participate in the understanding of the equilibrium between the host genome and its TEs.
Material and Methods
Drosophila stocks
Drosophila strains were maintained in the laboratory at 24° as small-mass cultures. Chicharo, Mayotte, Makindu and Zimbabwe are wild-type strains, anciently sampled from the field; w501 is the major strain of the D. simulans sequenced genome.
Small RNA analysis
We used small RNA-seq data obtained from ovaries from Akkouche et al. (2013) and Lerat et al. (2017), corresponding to the Chicharo, Mayotte and Makindu strains. We first removed adapter sequences using cutadatp (Martin 2011) -a CTGTAGGCACCATCAA. Using PRINSEQ lite version 0.20.4 (Schmieder and Edwards 2011), we filtered reads of size 23 to 30 nt and considered these as piRNAs. Using a modified version of the TEcount module of TEtools (Lerat et al. 2017), we mapped the reads against a list of TE sequences either in the sense or antisense directions. The list of TE sequences is made of the TE insertions retrieved from D. simulans sequenced genome (Lerat et al. 2017). Read count numbers were normalized to miRNA read count numbers (miRNA sequences retrieved from FlyBase: dsim-all-miRNA-r2.02.fasta.gz).
We looked for ping-pong signatures using signature.py with the options min_size = 23 and max_size = 30 (Antoniewski 2014). We used as input files SAM alignments obtained from cleaned small RNA reads against our C-subfamily complete (8.5 kb) reference tirant sequence: accession number AC0054444, positions 50,203 to 58,729, extracted from the D. melanogaster genome, as already described in Fablet et al. (2006). We used bowtie (–best) (Langmead et al. 2009). We analyzed base composition at each position for piRNAs aligned against tirant using SAMStat (Lassmann et al. 2011).
To visualize read mapping along tirant sequence, we sampled 40,000,000 reads from each small RNA-seq sample using the fastq-sample program from fastq-tools 0.8 (https://homes.cs.washington.edu/∼dcjones/fastq-tools/), and then we selected 23-30 nt-long reads as mentioned above. We used bowtie (–best) (Langmead et al. 2009) to map these small RNA-seq reads against our C-subfamily reference tirant sequence (see above). We removed the 3′ LTR, so that reads corresponding to LTRs should map at a unique location. We filtered out alignments with a mapq score below 20 using samtools (Li et al. 2009). Visualization of the alignments was performed using BamView (Carver et al. 2010).
Cell culture and reporter gene assays
We used reporter gene assays to test the sense and antisense promoter potentials of the LTRs of both subfamilies. LTR sequences were PCR amplified from the Mayotte strain (see Supplementary Material S1 for primer sequences). Primers were designed to include KpnI and HindIII restriction sites, to allow subsequent directional cloning into the pGL4.10 plasmid (Promega, luciferase reporter gene) using T4 DNA ligase (NEB). S2 Drosophila cells were transfected using Cellfectin (Invitrogen) and lyzed for analysis after 24 h of incubation (6 replicates per construct). Luminescence was then estimated using the Dual-Luciferase Reporter Assay System (Promega).
Genome sequencing and assemblies
Genomic DNA samples were treated with RNAse A at 37° for 30 min and purified using Phenol:Chloroform extraction. The purified genomic DNA samples were fragmented using Covaris S220 sonicator to get fragments of size 300-800 bp. Sequencing libraries were prepared from these fragmented DNA samples using NEBNext UltraTM DNA Library Prep Kit (Illumina, # E7370L). All three libraries were sequenced in one lane of an Illumina HiSeqV4 with 125 bp paired-end reads at the Next Generation Sequencing unit of the Vienna Biocenter Core Facilities (VBCF http://vbcf.ac.at).
Approximately 100 million read pairs (24 Gb) from three accessions (Mayotte, Makindu and Chicharo) were obtained and raw reads were filtered using the unsupervised quality trimming program UrQt (Modolo and Lerat 2015). Contigs were then generated by the Ray assembler v2.3.1 (Boisvert et al. 2010) using kmer length varying from 25 to 45. Best assemblies were selected by maximizing both NG50 and the number of contigs larger than 10 kb. For each accession, the assembled genome size was over 100% of the expected genome size (146.7 Mb). The final contig NG50 were 5.38 kb (k = 43), 11.89 kb (k = 39) and 7.36 kb (k = 35), with maximum scaffold length of ∼130 kb, 495 kb and 495 kb for Mayotte, Makindu and Chicharo, respectively. In order to assess completeness of genome assemblies, we searched for conserved genes across arthropods using BUSCO (Simão et al. 2015). Among the set of 2,675 genes, 2,254 (84%), 2,403 (90%) and 2,269 (85%) were recovered as complete and single copy in the assemblies of Mayotte, Makindu and Chicharo, respectively.
Genome and transcriptome analyses
We ran RepeatMasker (Smit et al. 2013) on the above assemblies (default parameters, -species Drosophila) and retrieved “Tirant” accessions in the output file in order to identify tirant insertion sites. Then, we extracted the flanking sequences for the insertions found above and located them on droSim1 assembly using the BLAT tool (Kent 2002) of UCSC Genome Browser (http://genome.ucsc.edu). We checked that shared tirant insertions had the same breakpoints in all strains.
RNA-seq data from ovaries and the corresponding computed read counts for all TE families, including tirant, were obtained from Lerat et al. (2017) for five strains (Chicharo, Makindu, Mayotte, Zimbabwe, and w501). Differential expression analysis was performed using DESeq2 (Love et al. 2014), which model internally corrects for library size, and provides normalization. Read counts obtained for genes on one hand and TEs on the other hand (Lerat et al. 2017) were concatenated to make the complete read count table that was analyzed at once using DESeq2.
The tkv gene had no assigned D. simulans ortholog in the FlyBase gene list that we used for RNA-seq analysis at that time (Lerat et al. 2017). In 2018, FBgn0194057 has been assigned as D. simulans tkv ortholog (OrthoDB v9.1). Thus, we retrieved the FBgn0194057 sequence, performed the same procedure as for the other genes (Lerat et al. 2017), and added the corresponding read counts to our count table.
To visualize read mapping along tirant sequence, we sampled 90,000,000 reads from each DNA-seq sample and 25,000,000 reads from each RNA-seq sample (one biological replicate from each strain) to ensure even abundance of reads. In addition, for RNA-seq data, we only used reads longer than 20 nt (PRINSEQ filter, as described above). We used the fastq-sample program from fastq-tools 0.8 (https://homes.cs.washington.edu/∼dcjones/fastq-tools/) and the fastq_sampler.py script (Modolo 2018). Then, we used bowtie2 (–very-sensitive) (Langmead and Salzberg 2012) and mapped DNA-seq and RNA-seq reads against our C-subfamily reference tirant sequence (see above). We removed the 3′ LTR, so that reads corresponding to LTRs should map at a unique location. We filtered out alignments with a mapq score below 20 using samtools (Li et al. 2009). Visualization of the alignments was performed using BamView (Carver et al. 2010).
Transcript structure analysis: we used apytram (apytram v1.1 (Rey et al. 2017)) to assemble independently Hs6st and tkv in Chicharo, Makindu and Mayotte RNA-seq samples (replicates #1). Apytram is an implementation of the Target Restricted Assembly Method (Johnson et al. 2013). This method allows to focus on the assembly of only one gene of a transcriptome and not the whole transcriptome. It is based on an iterative process: at the first iteration, a reference gene from another species is used as bait sequences to fish reads in RNA-seq data using BLAST (Camacho et al. 2009). Paired reads are used to enlarge this batch of reads. Reads are then de novo assembled using Trinity (Grabherr et al. 2011), and a new iteration begins with the reconstructed sequences as baits. Here, we used the Hs6st and tkv genes from D. melanogaster as references. This iterative process allows to reconstruct transcripts step by step from homologous regions between D. melanogaster and D. simulans, and thus non homologous regions can be reconstructed in D. simulans transcripts. In addition, if ever tirant is exonized in one of the strains, apytram will be able to catch it. Then, to analyze Hs6st and tkv transcript structures in our samples, we aligned output transcripts on D. simulans genome (ASM75419v3.41) using exonerate (Slater and Birney 2005). Finally, we manually checked the hit distribution against D. simulans genome to search for the presence of tirant in transcript sequences. We used TopHat2 (Kim et al. 2013) and ggsashimi (Garrido-Martín et al. 2018) to view transcript structures using sashimi plots.
ChIP assays
Biological duplicates were produced. For each replicate, seventy ovary pairs were dissected from three to five day-old females and flash frozen to maintain chromatin integrity. Chromatin extraction and sonication were carried out as described in Akkouche et al. (2013) with the following modifications: 30 sonication cycles (30 sec on and 30 sec off on high) were performed to obtain chromatin fragments comprised between 200 and 500 bp. The chromatin fragments were then aliquoted and diluted into six IP samples of 500 µL along with a 10% Input sample. IP samples were then immunoprecipitated following the manufactor’s protocol (Magna ChIP, Millipore) with 3 µg of antibodies (H3K4me3: Abcam # ab8580; H3K9me3: Actif Motif # 39161).
Quantitative PCR (qPCR) was used to estimate IP enrichment as previously described (Fablet et al. 2009; Akkouche et al. 2013) and performed as technical triplicates. For each of the studied tirant insertions, we used qPCR primers located at the insertion site and ∼2 kb away, in both directions. At the insertion site, we designed primer pairs displaying one primer within tirant and the other primer in the flanking sequence. For strains devoid of tirant insertion, we used the same primer in the flanking region as mentioned above and designed a second primer in the flanking region beyond the site of tirant insertion. These two primer pairs allow us to amplify tirant insertion sites in strains where tirant is present as well as in strains were tirant is absent. Primer sequences are provided in Supplementary Material S1. As controls, we used light for H3K9me3 enrichment (Fw: 5′-GCT AGG CAA TGA CAA AGT CCT TTG GG-3′ and Rv: 5′-GCA TTC GTC TGA AGT CGG CAG ATA G-3′), and rpl32 for H3K4me3 enrichment (Fw: 5′-CAG CTT CAA GAT GAC CAT C-3′ and Rv: 5′-GTT CGA TCC GTA ACC GAT GT-3′). Raw data from qPCRs are provided in Supplementary Material S2.
Long PCR
We PCR amplified tirant insertion at the Hs6st locus using DreamTaq (Fermentas), with a 7 min elongation time and a 68° hybridization. Primer sequences are 5′- GCAACACTGACAGCAACTACA -3′ and 5′- TCCTTGCTAGCTACATGGAAC -3′.
Data Availability
Genomic raw reads are available under the SRA accession number SRP128969. Genome assemblies, modified versions of TEcount.py and the list of TE sequences are available at ftp://pbil.univ-lyon1.fr/pub/datasets/Fablet2018. SupplMat S1 contains primer sequences. SupplMat S2 contains ChIP qPCR results. SupplMat S3 is an alignment of reference C and S sequences. Alignments of piRNAs against tirant reference sequence are provided Figure S1. Figure S2 displays graphics of piRNA base composition. Gels obtained from PCR products are shown Figure S3. Sashimi plots for tkv and Hs6st splicing are provided Figure S4. Supplemental material available at Figshare: https://doi.org/10.25387/g3.7578380.
Results
Tirant is differentially expressed across wild-type strains
Using RNA-seq data obtained from ovary samples, we were able to quantify tirant transcript levels in D. simulans strains (Figure 1). As expected (Fablet et al. 2006, 2009), subfamily S transcript levels were found at background level in all strains, and so were subfamily C transcript levels in w501, Chicharo and Zimbabwe strains. Tirant produces significant amounts of transcripts only in Makindu and Mayotte strains (Figure 1A). The mapping of tirant reads against the reference sequence shows enrichment in the env region (Figure 1B). This pattern could reflect either the existence of a master copy deleted for gag and pol, or the prevalence of splicing events of the gag and pol ORFs, as described by Marsano et al. (2000) in D. melanogaster. The higher abundance of env reads in the Mayotte strain is in agreement with the production of the tirant Env protein in this strain, which we previously observed using immunostaining (Akkouche et al. 2012).
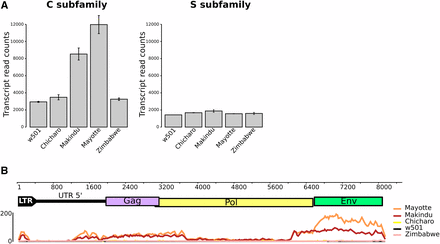
Tirant trancripts analyzed from RNA-seq data. A. Tirant transcript normalized read counts. B. Mapping of tirant reads against the reference tirant copy (subfamily C). The upper part is tirant structure, to scale (in bp). The lower part is read coverage along tirant sequence, obtained from samples of 25,000,000 paired-end reads. We removed LTR 3′ to get rid of multi-mapping issues.
Tirant displays specific sense and antisense piRNAs
We specifically analyzed small RNAs for the two strains that show significant tirant transcript levels (i.e., Makindu and Mayotte), and one strain presenting background levels (i.e., Chicharo). We found sense and antisense tirant-specific piRNAs, corresponding to both the C and S subfamilies. Sequence similarity between subfamilies C and S varies along tirant sequence: it is the lowest in the UTR 5′ region (∼65% (Fablet et al. 2006)) and displays higher values within coding sequences (76–85% global similarity rate (Fablet et al. 2009)). Although we cannot exclude that some reads may be mis-attributed to the other subfamily, it should not happen for the majority. Except for a peak in the LTR region, tirant piRNAs evenly map along tirant reference sequence in all three strains (Figure S1). Tirant piRNAs displayed comparable proportions of antisense piRNAs to total piRNAs in all three strains and tirant subfamilies, ranging from 0.798 to 0.869 (Figure 2A). In addition, we looked for ping-pong signatures, which are 10 nt overlaps between sense and antisense piRNAs (Brennecke et al. 2007). We found clear, similar enrichments for 10 nt overlaps in all three strains (Figure 2C), indicative of similar, functional, secondary piRNA pathways against tirant in all three strains. In addition, these piRNAs showed expected enrichment in 1U and 10A (Figure S2) (Saito et al. 2006; Brennecke et al. 2007).
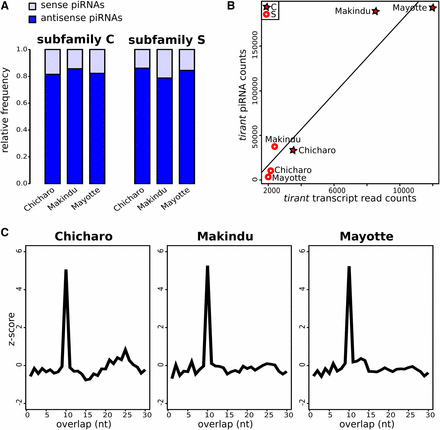
Tirant-specific piRNAs. A. Relative proportions of sense and antisense piRNAs. B. Positive correlation between tirant transcript normalized read counts and tirant piRNA read counts in wild-type strains. C. Ping-pong signatures: there is a clear enrichment in 10 nt overlaps.
Moreover, our data allowed us to reveal a positive correlation between tirant piRNA read counts and tirant transcript read counts (Pearson correlation, r = 0.963, p-value = 0.002, Figure 2B). The correlation remains strong using only sense (Pearson correlation, r = 0.965, p-value = 0.002), or antisense (Pearson correlation, r = 0.946, p-value = 0.004) piRNAs.
Tirant LTRs display antisense promoter activity
As new TE insertions may behave as dual-strand piRNA clusters (Mohn et al. 2014; Shpiz et al. 2014), we wondered whether tirant copies could be able to produce antisense piRNA precursors. To investigate such hypothesis, we tested the C and S LTRs for their sense and antisense promoter abilities. We known from a previous work that sequence similarity between C and S LTRs is 65% on average, while sequence similarity within each subfamily is higher than 98% (Fablet et al. 2006). It appears that the different constructs behave significantly differently regarding gene expression promotion (Kruskal Wallis test, df = 3, p-value = 2.10−4), with LTR C sense being the strongest promoter and LTR S sense being the weakest. In addition, we find that both subfamily LTRs display weak antisense promoter activity (Figure 3) (Wilcoxon tests, C antisense vs. S sense: p-value = 0.002, S antisense vs. S sense: p-value = 0.002, C antisense vs. S antisense: p-value = 0.485). Therefore, the results suggest that, although subfamily S is not significantly involved in the production of tirant transcripts, and albeit the weak activity observed in vitro, it may rather be involved in the production of antisense piRNA precursors.
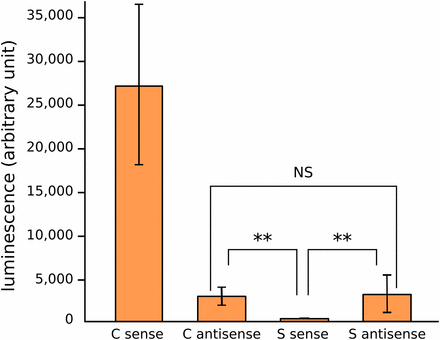
Tirant LTR promoter activity. LTRs of C and S subfamilies in sense and antisense orientations were cloned upstream of a luciferase reporter gene and tested for promoter activity in S2 cells.
Tirant displays insertional polymorphism
The influence of a TE on the host genome depends on its insertion sites. To investigate tirant’s impacts, we produced genome assemblies for Chicharo, Makindu and Mayotte, and used RepeatMasker (Smit et al. 2013) to retrieve tirant insertion sites. We excluded scaffolds exclusively made of tirant. Nested tirant insertions into other TE sequences were also excluded since we are not able to locate them precisely on the genome. The final number of retrieved tirant insertions is congruent with our knowledge of tirant copy numbers (Fablet et al. 2006, 2009; Akkouche et al. 2012), indicating that we did not missed a significant number of loci.
Overall, we localized precisely 15 insertions, the majority of which are unique to one strain (Table 1). Among these, six are located in introns and two upstream transcription start sites (TSSs), which are the insertions the most likely to have an impact on genes. Insertion #10 is located 242 bp upstream of CanA1 TSS, and insertion #11 533 bp upstream of Lamp1 TSS. We identified five additional insertions, attributed to the U chromosome (Unknown chromosome), and which likely correspond to heterochromatic regions. Among these, one is shared across strains.
Tirant insertion sites as identified using RepeatMasker on genome assemblies. +: insertion found in the genome assembly of the strain. Due to the fragmented nature of our assemblies, we are not able to interpret “absence of detection” as “absence of tirant”. These data do not distinguish tirant subfamilies. TSS: Transcription Start Site. U: Unkown chromosome. ≥ LTR: we detected one LTR (417 bp) at the extremity of a scaffold, which means that we do not know whether the insertion is a solo LTR or longer. Insertion #10 is located 242 bp upstream of CanA1 TSS, and insertion #11 533 bp upstream of Lamp1 TSS. D. simulans orthologs as assigned in OrthoDB v9.1 (retrieved via FlyBase in november 2018) are mentioned in brackets and italics.
| Insertion # | Chr | Annotation | Insertion size (bp) | Mayotte | Makindu | Chicharo |
|---|---|---|---|---|---|---|
| 1 | 3R | Intron CG34383 (FBgn0190438) | 41 | + | + | + |
| 2 | 3L | 102 | + | |||
| 3 | 3R | Intron Hs6st (FBgn0041661) | ≥ LTR | + | ||
| 4 | 2L | Intron vri (FBgn0194720) | ≥ LTR | + | ||
| 5 | 3R | Intron CG11873 (NA) | ≥ LTR | + | ||
| 6 | 3R | > 95 | + | |||
| 7 | 2L | 446 | + | + | ||
| 8 | 3h | 490 | + | |||
| 9 | 2h | > 589 | + | |||
| 10 | 3R | Near CanA1 TSS (FBgn0192983) | > 96 | + | ||
| 11 | 2L | Near Lamp1 TSS (FBgn0195671) | 704 | + | + | + |
| 12 | 2L | Intron tkv (FBgn0194057) | ≥ LTR | + | ||
| 13 | 2L | > 100 | + | |||
| 14 | 2R | > 95 | + | |||
| 15 | 2R | Intron CG17684 (FBgn0268563) | 8014 | + | ||
| 16 | U | > 99 | + | + | + | |
| 17 | U | >98 | + | + | ||
| 18 | U | 115 | + | |||
| 19 | U | > 93 | + | |||
| 20 | U | 438 | + |
| Insertion # | Chr | Annotation | Insertion size (bp) | Mayotte | Makindu | Chicharo |
|---|---|---|---|---|---|---|
| 1 | 3R | Intron CG34383 (FBgn0190438) | 41 | + | + | + |
| 2 | 3L | 102 | + | |||
| 3 | 3R | Intron Hs6st (FBgn0041661) | ≥ LTR | + | ||
| 4 | 2L | Intron vri (FBgn0194720) | ≥ LTR | + | ||
| 5 | 3R | Intron CG11873 (NA) | ≥ LTR | + | ||
| 6 | 3R | > 95 | + | |||
| 7 | 2L | 446 | + | + | ||
| 8 | 3h | 490 | + | |||
| 9 | 2h | > 589 | + | |||
| 10 | 3R | Near CanA1 TSS (FBgn0192983) | > 96 | + | ||
| 11 | 2L | Near Lamp1 TSS (FBgn0195671) | 704 | + | + | + |
| 12 | 2L | Intron tkv (FBgn0194057) | ≥ LTR | + | ||
| 13 | 2L | > 100 | + | |||
| 14 | 2R | > 95 | + | |||
| 15 | 2R | Intron CG17684 (FBgn0268563) | 8014 | + | ||
| 16 | U | > 99 | + | + | + | |
| 17 | U | >98 | + | + | ||
| 18 | U | 115 | + | |||
| 19 | U | > 93 | + | |||
| 20 | U | 438 | + |
| Insertion # | Chr | Annotation | Insertion size (bp) | Mayotte | Makindu | Chicharo |
|---|---|---|---|---|---|---|
| 1 | 3R | Intron CG34383 (FBgn0190438) | 41 | + | + | + |
| 2 | 3L | 102 | + | |||
| 3 | 3R | Intron Hs6st (FBgn0041661) | ≥ LTR | + | ||
| 4 | 2L | Intron vri (FBgn0194720) | ≥ LTR | + | ||
| 5 | 3R | Intron CG11873 (NA) | ≥ LTR | + | ||
| 6 | 3R | > 95 | + | |||
| 7 | 2L | 446 | + | + | ||
| 8 | 3h | 490 | + | |||
| 9 | 2h | > 589 | + | |||
| 10 | 3R | Near CanA1 TSS (FBgn0192983) | > 96 | + | ||
| 11 | 2L | Near Lamp1 TSS (FBgn0195671) | 704 | + | + | + |
| 12 | 2L | Intron tkv (FBgn0194057) | ≥ LTR | + | ||
| 13 | 2L | > 100 | + | |||
| 14 | 2R | > 95 | + | |||
| 15 | 2R | Intron CG17684 (FBgn0268563) | 8014 | + | ||
| 16 | U | > 99 | + | + | + | |
| 17 | U | >98 | + | + | ||
| 18 | U | 115 | + | |||
| 19 | U | > 93 | + | |||
| 20 | U | 438 | + |
| Insertion # | Chr | Annotation | Insertion size (bp) | Mayotte | Makindu | Chicharo |
|---|---|---|---|---|---|---|
| 1 | 3R | Intron CG34383 (FBgn0190438) | 41 | + | + | + |
| 2 | 3L | 102 | + | |||
| 3 | 3R | Intron Hs6st (FBgn0041661) | ≥ LTR | + | ||
| 4 | 2L | Intron vri (FBgn0194720) | ≥ LTR | + | ||
| 5 | 3R | Intron CG11873 (NA) | ≥ LTR | + | ||
| 6 | 3R | > 95 | + | |||
| 7 | 2L | 446 | + | + | ||
| 8 | 3h | 490 | + | |||
| 9 | 2h | > 589 | + | |||
| 10 | 3R | Near CanA1 TSS (FBgn0192983) | > 96 | + | ||
| 11 | 2L | Near Lamp1 TSS (FBgn0195671) | 704 | + | + | + |
| 12 | 2L | Intron tkv (FBgn0194057) | ≥ LTR | + | ||
| 13 | 2L | > 100 | + | |||
| 14 | 2R | > 95 | + | |||
| 15 | 2R | Intron CG17684 (FBgn0268563) | 8014 | + | ||
| 16 | U | > 99 | + | + | + | |
| 17 | U | >98 | + | + | ||
| 18 | U | 115 | + | |||
| 19 | U | > 93 | + | |||
| 20 | U | 438 | + |
It is to be noted that, following this procedure, the tirant insertion into the tkv gene (insertion #12) is only found in the Makindu genome, while we have experimental evidence that it also exists in the Mayotte strain. This stresses on the non exhaustive nature of this approach, which strongly relies on scaffold assemblies. Indeed, we used a PCR approach to confirm tirant insertion polymorphism for insertions #3 and #12, which are studied in more details below. (See Supplementary Material S1 for primer sequences). For insertion #3, PCR profiles were congruent with tirant being present only in Mayotte and absent in Makindu and Chicharo (Figure S3A). For insertion #12, PCR profiles indicated tirant presence in Makindu and Mayotte, and tirant absence in Chicharo (Figure S3B). However, we could also detect a very faint band indicating tirant absence in Makindu. This suggests that this insertion may be heterozygous or even absent in few individuals.
Accessing the precise structure of each tirant insertion is difficult without long reads. Therefore, we mapped genomic reads against our tirant reference sequence to have an idea of the representativeness of each region within the genomes of our strains. We found that reads map evenly along the reference sequence for Chicharo and Makindu, suggesting the presence of full-length copies (Figure 4). Mayotte displays the highest number of reads after normalization, in agreement with a higher number of tirant copies. We note a slight depression in the center of the pol and env ORFs (Figure 4), suggesting the existence of internally deleted copies. The drop of mapping coverage within the 5′ UTR region is due to our mapq filter (see Material and Methods), which prevents multi-mapping against repeated regions, such as the minisatellite that is described within tirant 5′ UTR (Fablet et al. 2006).
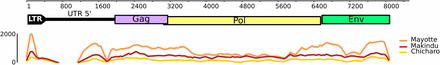
Mapping of genomic reads against tirant reference sequence. The upper part represents tirant structure, to scale (in bp). The lower part depicts read coverage along tirant sequence (subfamily C), obtained from samples of 90,000,000 paired-end reads. We removed LTR 3′ to get rid of multi-mapping issues.
Mild impact of tirant on neighboring genes
For those insertions located near or within genes (within introns), we tested whether tirant could have an effect on gene expression through modifications of chromatin structure. To do so, we used data produced from ovaries, and we focused on the insertions of tirant that fulfill the following criteria: i) the gene into which tirant is inserted displays a high enough number of reads in our RNA-seq dataset (> 100 reads), ii) its expression pattern is contrasted across strains with and without the tirant insertion (Lerat et al. 2017). We found only two of such insertions (Figure 5). The two insertions of interest are located within the Hs6st and tkv genes, respectively (insertions #3 –only in Mayotte– and #12 –in Mayotte and Makindu– in Table 1, respectively). In both cases, gene transcript levels are lower for strains where tirant is present compared to strains devoid of tirant at the considered locus (DESeq2 padj values: Hs6st: Chicharo vs. Mayotte: 0.0157, Makindu vs. Mayotte: 0.0916; tkv: Chicharo vs. Makindu: 0.0617, Chicharo vs. Mayotte: 2.10−8). Moreover, FlyBase D. simulans genome annotation indicates the presence of additional genes within Hs6st (FBgn0190828, FBgn0270486, FBgn0190826, and FBgn0269631) and tkv (FBgn0270637, FBgn0194717, FBgn0270509, and FBgn0194718) introns. However, virtually none of the reads of our data sets map against these sequences.
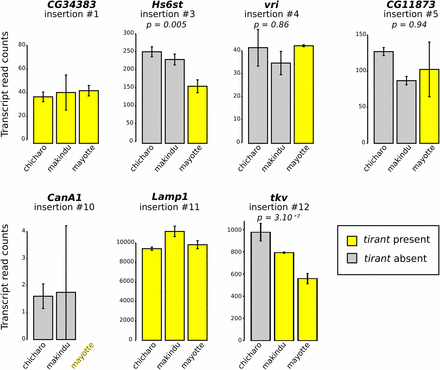
RNA-seq results for genes at or near tirant insertions. Normalized read counts. Please refer to Table 1 for tirant insertion details. Error bars are standard deviations on biological duplicates. When tirant insertion is polymorphic, we provide adjusted p-values comparing strains with and without tirant insertion, as calculated by DESeq2. Gene CG17684 (insertion #15) has no D. simulans ortholog in the list of genes used for RNA-seq data mapping.
We investigated chromatin structure to look for epigenetic modifications that could spread on neighboring regions. We performed ChIP experiments on H3K4me3 and H3K9me3. The former is characteristic of open chromatin conformation, particularly found at promoters; the later is the heterochromatic mark known to be involved in TE transcriptional silencing. We used qPCR primers located at the insertion site and ∼2 kb away, in both directions (Figure 6A, 6B). Primers at the insertion sites were the same as those mentioned above to detect tirant presence (Supplementary Material S1). No obvious effect of tirant presence on either chromatin mark at the Hs6st gene is observed: Mayotte enrichment levels (tirant present) lie in between Chicharo and Makindu levels (tirant absent). On the contrary to Hs6st insertion, it appears that tirant neighborhood is moderately enriched in H3K9me3 at the tkv insertion site compared to strains without tirant (fold change = 1.43 ; t-test, p-value = 0.098). In addition, these histone post-translation modifications spread at least 2 kb away downstream tirant (fold change = 1.41 ;t-test, p-value = 0.002). Regarding H3K4me3 enrichments, we could not detect differences between strains with and without tirant at the tkv locus.
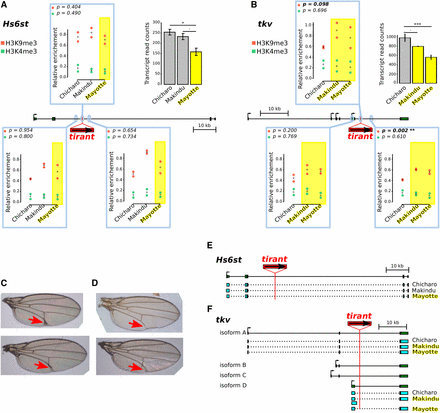
Analysis of two tirant insertions within introns. A. and B. Histone mark enrichments at two tirant insertion sites. P-values in the upper left corners are produced from t-tests between strains with the tirant insertion (yellow) and strains without tirant (uncolored). Left panel: A. tirant insertion #3 into Hs6st, right panel B.: tirant insertion #12 into tkv. Genes are drawn to scale in the middle of each panel; thin lines: introns, thick boxes: exons. H3K4me3 and H3K9me3 enrichments are shown in green and red, respectively, and quantified relative to control genes (see Material and Methods section). They are displayed in boxes corresponding to the location of the qPCR amplicons: either at the tirant insertion site or 2 kb away. Biological duplicates were produced for ChIP experiments. “+” indicates the mean between biological replicates. Normalized transcript levels for the considered genes are provided in the upper right corners (as already shown Figure 5). Error bars are standard deviations. Strains in which tirant is present at the considered locus are highlighted in yellow. Statistical significance for RNA-seq results was assessed by padj values provided by the DESeq2 analysis. Padj values: Hs6st: Chicharo vs. Mayotte: 0.0157, Makindu vs. Mayotte: 0.0916, tkv: Chicharo vs. Makindu: 0.0617, Chicharo vs. Mayotte: 2.10−8. C. and D. Vein phenotype in the Mayotte strain. C. We observed thick L5 veins (red arrows) in seven out of 97 flies in the Mayotte strain. D. Wild-type vein phenotype observed in 90 out of 97 flies in Mayotte (the same phenotype is also observed in all examined flies in Makindu and Chicharo). E. and F. Transcript structures. Reference structures from D. melanogaster, as retrieved from the UCSC Genome Browser, are depicted in dark green, thin lines: introns, thick boxes: exons. Reconstructed exons are shown in light blue. E. Hs6st transcript structures. All three strains display the same transcript structure as described in D. melanogaster. F. tkv transcript structures. We observe a Makindu-specific transcript of isoform D.
Since tkv mutations are known to alter wing vein phenotypes (Lindsley and Zimm 1992), we examined wing morphologies in our flies (we did not separate sexes). We found seven flies out of 97 in the Mayotte strain which displayed thick L5 veins (Figure 6C, 6D), while we did not find any out of 100 Makindu flies nor 100 Chicharo flies (Fisher exact test, p-value = 0.014). It is to be noted that Mayotte is the strain that shows the strongest reduction in tkv transcript levels.
As it was recently shown that piRNAs may also regulate TEs through splicing (Teixeira et al. 2017), we analyzed transcript structures in Hs6st and tkv. Hs6st displays virtually no transcript isoform variation in the reference D. melanogaster genome. Unsurprisingly, we found the described Hs6st transcript to be produced in all three strains (Figure 6E). In contrast, four tkv isoforms are described in D. melanogaster. Isoforms B and C are not found to be produced in our samples, and isoform A displays the same reference structure in all three strains. Variability was observed regarding isoform D: the first exon is 1,371 bp longer in the Makindu strain compared to the others (Figure 6F). Sashimi plots are provided Figure S4. Interestingly, this variable splice site is located less than 800 bp from tirant insertion in Makindu and Mayotte, and thus in the region where we find chromatin structure to be modified in association with tirant insertion. It is thus tempting to speculate that tirant-induced chromatin modifications may affect splice site definition. In addition, we note that we do not find evidence of tirant exonization neither in Hs6st nor in tkv.
Discussion
The repertoire of tirant-specific piRNAs
Our in-depth analysis of tirant-specific small RNAs revealed the presence of both sense and antisense piRNAs. While the primary piRNA pathway exclusively produces antisense piRNAs, the secondary piRNA pathway leads to the production of both sense and antisense piRNAs, characterized by the so-called ping-pong signature (Brennecke et al. 2007). Thus, the detection of sense tirant piRNAs indicates that the secondary pathway is involved in tirant control, as already proposed by previous experimental analyses (Akkouche et al. 2013). This is reinforced by the identification of ping-pong signatures, which are similar across strains. However, we cannot exclude the involvement of the primary pathway as well, especially considering the high proportions of antisense tirant-specific piRNAs.
So far, we do not know which piRNA clusters are involved in the production of tirant-specific piRNAs in D. simulans strains; however, our observation of antisense promoter properties for both subfamilies, albeit weak, indicates that some copies may have the ability to behave as dual-strand piRNA clusters, as proposed by two research studies (Mohn et al. 2014; Shpiz et al. 2014). While the S subfamily is not transcribed and apparently not involved in tirant activity, this potential role in piRNA precursor production may also explain why it is found to be conserved across strains (Fablet et al. 2006). However, although we provide in vitro evidence that some tirant LTR sequences may behave as antisense promoters, we do not know whether the antisense tirant piRNAs that we detect are indeed produced from these promoters or from promoters outside of the element itself.
Moreover, while we observed significant differences in tirant piRNA amounts across strains, we could not detect differences in sense vs. antisense piRNA ratios neither in ping-pong signatures, indicating that tirant control by the piRNA pathway may take place the same way and with comparable efficiencies in the three considered strains.
Dynamics of tirant control by piRNAs
Our survey in wild-type strains revealed a positive correlation between the amounts of tirant transcripts and tirant piRNAs in ovaries in normal conditions. This is in agreement with what we found using all TE families in the same strains (Lerat et al. 2017). This is also congruent with data obtained by others from D. melanogaster laboratory strains in normal conditions (Kelleher and Barbash 2013), and in accordance with the accepted model claiming that TE transcripts fuel the ping-pong loop (Senti et al. 2015).
Nevertheless, these results contrast with what was found in other studies. Notably, in a previous work, in order to understand tirant control, we performed crosses between Makindu and Chicharo, because they display contrasted tirant copy numbers and activities (Akkouche et al. 2013). When the mother comes from Makindu and the father comes from Chicharo, tirant is properly controlled in the progeny. This direction of cross is called “RT” (Regulated Tirant). However, when the reciprocal cross is performed, tirant shows a strong accumulation of transcripts in the somatic follicle cells of the progeny’s ovaries (Akkouche et al. 2013). This direction of cross is called “NRT” (Non-Regulated Tirant). This disruption of tirant control is associated with a lack of tirant-specific piRNAs, while these piRNAs remain relatively abundant in the RT direction of cross (Akkouche et al. 2013). It results in a negative correlation between tirant transcript amounts and tirant-specific piRNA amounts.
We propose that the nature of the correlation between TE transcripts and piRNAs reflects genome stability. In normal conditions – which corresponds to a balanced relationship between the genome and its TEs, the correlation is positive. When the equilibrium is broken – and so far we do not know which sensor could detect such event –, the correlation becomes negative. Such patterns were also observed in the case of the I non-LTR retrotransposon in D. melanogaster strains (Chambeyron et al. 2008). Indeed, Chambeyron et al. described that strain JA has no I transcripts nor I-specific piRNAs while strain HT2 displays high amounts of both I transcripts and piRNAs (Chambeyron et al. 2008). This corresponds to the positive correlation we propose at equilibrium. On the other hand, when dysgenic crosses are performed and disrupt genome / I stable relationship, an opposite correlation is observed: SF females –which suffer from sterility– accumulate I transcripts due to a lack of I piRNAs; on the contrary, RSF females –in which fertility is restored– lower I transcript levels due to a high production of I piRNAs. Thus, the results obtained from tirant in the present work may reflect a behavior common to all TE families.
Tirant’s soft influence on neighboring genes
In most organisms, TE control is mainly achieved by epigenetic mechanisms (Slotkin and Martienssen 2007; Siomi et al. 2011). In D. melanogaster, these essentially consist in piRNAs and H3K9me3 (Saito et al. 2006; Aravin et al. 2007; Sienski et al. 2012; Le Thomas et al. 2013). We know that this heterochromatic mark may spread to flanking regions up to a 5 kb distance (Rebollo et al. 2011). Rebollo et al. used mouse strains and did not find that heterochromatin spreading recurrently affected gene activity, probably due to the fact that such TE insertions would be too deleterious (Rebollo et al. 2011). In a Drosophila system, Sienski and colleagues also observed such spreading, which in addition did impact neighboring genes in mutants in cell cultures (Sienski et al. 2012). In the present study, we used the tirant system to investigate the spreading of TE chromatin marks in a natural context. We found a large level of insertion polymorphism across the strains we investigated (Table 1). We identified two cases, corresponding to insertions into introns of the Hs6st and tkv genes, associated with changes in gene transcript levels. While we could not detect any effect of tirant insertion on chromatin structure in the case of Hs6st, tirant insertion into the tkv gene was associated with a moderate enrichment in H3K9me3 marks at the insertion site and up to 2 kb away from the insertion site, compared to the strain devoid of tirant. In addition, we noted that the tirant effect that we detect in Makindu may be underestimated since there may be heterozygosity or absence of the insertion in some individuals. We searched for other TEs in the genome assemblies of all strains in a ∼10 kb window around this particular tirant insertion, and could not find any. This suggests that the effects observed on chromatin structure and gene expression may be attributed to the insertion of tirant. Moreover, we observed a thick veins phenotype in 7% of Mayotte flies while we could not observe it in Makindu nor Chicharo flies. Mayotte is the strain displaying the strongest reduction in tkv transcript levels (Mayotte vs. Chicharo: log2FC = -0.80). Although we cannot exclude other differences in genetic backgrounds, we propose that the tirant insertion into tkv in Mayotte may be responsible for this thick veins phenotype, through tkv expression reduction induced by heterochromatin formation. Nevertheless, variation in tkv transcript levels is observed –probably associated with micro-environmental variability–, which may explain why most flies do not show the phenotype.
In addition, as it was recently shown that piRNAs may also regulate TEs through splicing (Teixeira et al. 2017), we analyzed transcript structures in Hs6st and tkv. We found no effect of tirant insertion in Hs6st transcript structures. This observation parallels the absence of tirant effect on Hs6st chromatin conformation. In contrast, we observed variation in tkv splice sites nearby tirant insertion in the Makindu strain: the first exon of isoform D is 1,371 bp longer than expected. It is tempting to speculate that tirant-induced chromatin modifications disrupt splice site definition; however, we do not find the same pattern in the Mayotte strain, which also displays tirant insertion. Further investigation is needed at a larger scale to determine the impacts of TEs on splice site definition.
Tirant insertions have either no or moderate effects on nearby gene expression. The insertion into tkv may have an impact on chromatin structure and gene expression whereas the tirant insertion into Hs6st does not. Our results are congruent with the recently published study of Lee and Karpen, although regarding a different histone mark. Based on a D. melanogaster genome-wide study, they report the spreading of TE-associated H3K9me2 marks on regions flanking the insertions and an effect on the expression level of flanking genes only for about half of the TE insertions (Lee and Karpen 2017). So far, we have not identified which properties of a TE insertion make it affect nearby genes or not. We know from a previous work that tirant is full length at the tkv locus in the Makindu strain (Fablet et al. 2009). We performed a long PCR at the Hs6st locus and observed a band congruent with a full-length insertion as well (Figure S3C). In addition, both tirant sequences are oriented the same way as the gene. We may note that the Hs6st locus displays insertion polymorphism for helitron N1_Dbi, which is found approximately 1 kb away from tirant in Chicharo and Mayotte but not in Makindu. Apart from this helitron and tirant, no other TE sequences are found within a 4 kb window around tirant insertion site. To sum up, we still do not know why tirant affects tkv but not Hs6st.
Tirant effects are of small size: on average, a 30% reduction in tkv transcript amounts and an increase of 33–42% in H3K9me3 enrichment. If it were not a small effect, we speculate that the insertion would be too deleterious and removed from the genome by natural selection. In addition, cases were also reported in which TE insertions had only a modest impact on neighboring genes in standard conditions, but significant effects under stress conditions (Naito et al. 2009).
Conclusion
In the present study, we use natural variability regarding the tirant ERV of D. simulans to identify potential impacts of TEs on the host genome. We show that, to a moderate extent, a tirant insertion may spread heterochromatin to flanking regions where genes lie, and be associated with transcript level reduction for the gene, with potential phenotypic impacts. Our results also illustrate that TE effects on the genome may not be as pervasive as recently proposed (Lee and Karpen 2017) since we detected them only for one insertion and not for the others. In addition, our data suggest the existence of a dynamic relationship between TE transcripts and the piRNAs that control them. This is illustrated by a positive correlation between these two variables in normal conditions, which is reverted to a negative correlation in the case of dysgenic crosses. We speculate this may happen when asymmetry is too high between ping-pong partners abundances, –ie maternally transmitted piRNAs on one hand and TE transcripts on the other hand, for the piRNA pathway to be efficient.
Taken together, our results participate in the understanding of the equilibrium between the host genome and its TEs. This study opens the way to the investigation of the transition from the equilibrium disruption to a restored balance between the genome and its TEs, which are fundamental aspects of the understanding of TE biology and genome stability.
Acknowledgments
This work was performed using the computing facilities of the CC LBBE/PRABI, the luminometer of the IUT Lyon 1 Biology Department, and the Vienna Biocenter Campus Facility.
We thank Justine Picarle, Sonia Martinez, Nabila Berabez and Anaïs Alquezar for technical assistance. We thank Philippe Veber, Matthieu Boulesteix, and attendees of the XXth French National Congress on Transposable Elements for fruitful discussions. We thank Labex ECOFECT.
Footnotes
Supplemental material available at Figshare: https://doi.org/10.25387/g3.7578380.
Communicating Editor: J. Birchler
Literature Cited
Martin, M., 2011 Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet.journal 17: 10–12.
Modolo, L., 2018 l-modolo/fastq_sampler: everything is working. Zenodo.
Rey, C., T. Lorin, M. Sémon, and B. Boussau, 2017 apytram v1.1. Zenodo.
Smit, A. F., R. Hubley, and P. Green, 2013 RepeatMasker Open-4.0. http://www.repeatmasker.org.
Author notes
Current address: Institute of Evolutionary Biology (CSIC-Universitat Pompeu Fabra), Barcelona 08003, Spain.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}