





Abstract
Genome structure characterization can contribute to a better understanding of processes such as adaptation, speciation, and karyotype evolution, and can provide useful information for refining genome assemblies. We studied the genome of an important North American boreal forest pest, the spruce budworm, Choristoneura fumiferana, through a combination of molecular cytogenetic analyses and construction of a high-density linkage map based on single nucleotide polymorphism (SNP) markers obtained through a genotyping-by-sequencing (GBS) approach. Cytogenetic analyses using fluorescence in situ hybridization methods confirmed the haploid chromosome number of n = 30 in both sexes of C. fumiferana and showed, for the first time, that this species has a WZ/ZZ sex chromosome system. Synteny analysis based on a comparison of the Bombyx mori genome and the C. fumiferana linkage map revealed the presence of a neo-Z chromosome in the latter species, as previously reported for other tortricid moths. In this neo-Z chromosome, we detected an ABC transporter C2 (ABCC2) gene that has been associated with insecticide resistance. Sex-linkage of the ABCC2 gene provides a genomic context favorable to selection and rapid spread of resistance against Bacillus thuringiensis serotype kurstaki (Btk), the main insecticide used in Canada to control spruce budworm populations. Ultimately, the linkage map we developed, which comprises 3586 SNP markers distributed over 30 linkage groups for a total length of 1720.41 cM, will be a valuable tool for refining our draft assembly of the spruce budworm genome.
The order Lepidoptera (moths and butterflies) is one of the most diverse taxa of insects, with at least 157,000 species described to date (Kristensen et al. 2007; van Nieukerken et al. 2011). Among these species, many have been extensively studied due to their pest status, whereas others have been used as experimental models in fields as diverse as physiology, genetics, evolution and ecology (Roe et al. 2010). For example, the diamondback moth, Plutella xylostella, has been used in many studies examining the development of insecticide resistance (You et al. 2013), while the squinting bush brown, Bicyclus anynana, whose wing color patterns vary depending on the season, has proven a valuable system to study the evolution of phenotypic plasticity (Brakefield et al. 2007). Genomic characteristics unique to Lepidoptera have also drawn much attention. Moths and butterflies have holokinetic chromosomes (i.e., chromosome lacking a primary constriction, the centromere; Marec et al. 2010; Sahara et al. 2012) and meiotic recombination is limited to the male sex (Suomalainen et al. 1973; Turner and Sheppard 1975; Traut 1977; Nokkala 1987; Traut et al. 2007). Moreover, lepidopteran chromosome numbers show a high degree of variability, ranging from n = 5 to n = 224-226 in the species examined (Marec et al. 2010; Lukhtanov 2015; Blackmon et al. 2017), with an average around the inferred ancestral number of n = 31 chromosomes (Suomalainen 1969; Lukhtanov 2000; Ahola et al. 2014; Yasukochi et al. 2016), found in ca. one-third of species (Suomalainen 1969; Robinson 1971; De Prins and Saitoh 2003; Blackmon et al. 2017). Interestingly, a high degree of gene synteny has been observed among homologous chromosomes across distantly related taxa (Pringle et al. 2007; Yasukochi et al. 2009; Baxter et al. 2011; Yoshido et al. 2011; Van’t Hof et al. 2013; Ahola et al. 2014), and the patterns of synteny described so far indicate that the numerically altered karyotypes evolved via fusion and/or fission events (Marec et al. 2010; Ahola et al. 2014; Šíchová et al. 2015). Finally, the order Lepidoptera, along with its sister group Trichoptera (caddisflies) and some tephritid fruit flies, is part of a restricted assemblage of insects in which the female is the heterogametic sex (ZW or Z0) (Bush 1966; Traut et al. 2007; Marec et al. 2010; Sahara et al. 2012; Blackmon et al. 2017).
The spruce budworm, Choristoneura fumiferana, is another lepidopteran species that has attracted much research attention. Belonging to the family Tortricidae, it is native to North America, and its larvae mainly feed on the foliage of spruces and true firs. One of the hallmarks of spruce budworm population dynamics is the development of severe outbreaks lasting ca. 10 years and recurring every 30 to 40 years (Jardon et al. 2003; Boulanger et al. 2012). During these outbreaks, intense defoliation typically results in major growth reductions, and several years of severe infestations can cause widespread tree mortality (MacLean 1985). While considered a very serious pest of North American spruce and fir stands, the spruce budworm is also seen as playing a positive, essential role in forest renewal (MacLean 2016). Since 2006, a new outbreak has gradually developed and spread over several regions of the province of Quebec, affecting 7.1 MHa as of 2017 (Ministère des forêts, de la faune et des parcs 2017). Due to its significant economic impact on the pulp and lumber industry across the Canadian boreal forest, the spruce budworm has been the subject of numerous studies and has imposed itself as a model for the study of insect population dynamics in North America (Pureswaran et al. 2016).
The spruce budworm has also been at the center of many studies examining various aspects of its biology, including its cytogenetic characteristics. It was reported that its karyotype consists of 30 pairs of chromosomes, including a pair of large chromosomes, presumably sex chromosomes (Ennis 1976; Harvey 1997). According to recent data, large sex chromosomes are a typical feature of most tortricids, with Z chromosomes having arisen from the fusion between an ancestral Z chromosome and an autosome corresponding to chromosome 15 of Bombyx mori (Nguyen et al. 2013; Šíchová et al. 2013). However, the occurrence of a similar fusion in C. fumiferana has yet to be confirmed. In addition, the structure and composition of the W chromosome varies greatly among lepidopteran species, and characterization of these features can provide novel insights into the formation and evolution of sex chromosomes in the Lepidoptera (Traut and Marec 1997; Yoshido et al. 2005; Vítková et al. 2007; Šíchová et al. 2013; Dalíková et al. 2017; Mongue et al. 2017). In C. fumiferana, composition of the W chromosome has not yet been examined.
In the present work, we developed a SNP-based high-density linkage map of C. fumiferana and used cytogenetic techniques to study the composition and structure of its genome. More specifically, (i) we confirmed the presence of 30 pairs of chromosomes in both sexes of this species, including a large pair of sex chromosomes, (ii) we used synteny analysis to confirm the existence of a neo-Z chromosome and (iii) we conducted cytogenetic characterization of the structure of the W chromosome. The present work was initiated as part of a C. fumiferana genome project, whose main goal is to generate a high-quality reference genome for this species. The high-density linkage map developed here will help to position scaffolds on chromosomes.
Materials and Methods
Karyotype analysis
Chromosome number for Choristoneura fumiferana was determined from mitotic metaphase cells stained by fluorescence in situ hybridization (FISH) using (TTAGG)n telomeric probe (tel-FISH), which enables identification of chromosome ends. Mitotic chromosomes were obtained from wing imaginal discs of 5th instar male and female larvae using the spreading technique described in Mediouni et al. (2004). The telomeric probe was generated by non-template PCR according to the protocol of Sahara et al. (1999) and labeled with Cy3-dUTP (Jena Bioscience, Jena, Germany) using a Nick Translation Kit (Abbott Molecular Inc., Des Plaines, USA) with 75 min incubation at 15°. FISH with the (TTAGG)n telomeric probe was performed following the protocol of Yoshido et al. (2005). The hybridization mixture contained 100 ng of telomeric probe and 25 µg of sonicated salmon sperm DNA (Sigma-Aldrich, St. Louis, USA) in 10 µL of 50% formamide and 10% dextran sulfate in 2 x SSC.
To identify the sex chromosomes of C. fumiferana, we used genomic in situ hybridization (GISH) combined with tel-FISH as described in Yoshido et al. (2005) and Šíchová et al. (2015). GISH is based on hybridization of a fluorescently labeled female genomic DNA (gDNA) in the presence of an excess of unlabeled competitor DNA (i.e., male gDNA). With this approach, the W chromosome is identified by strong binding of female labeled DNA due to its repetitive character. This approach has successfully identified the W chromosome in several lepidopteran species (Mediouni et al. 2004; Fuková et al. 2005; Yoshido et al. 2006).
Spread chromosome preparations were obtained from testes and ovaries of 5th instar larvae as described in Mediouni et al. (2004). For GISH, total gDNA from males and females was isolated using the CTAB method adapted from Winnepenninckx et al. (1993). Female gDNA was labeled with fluorescein-12-dUTP (Jena Bioscience) using the Nick Translation Kit (Abbott Molecular Inc.), with 4 h of incubation at 15°, and male gDNA was sonicated using a Sonopuls HD 2070 (Bandelin Electric, Berlin, Germany) and used as competitor DNA. For GISH combined with tel-FISH, the probe mixture contained fluorescein-labeled female gDNA (300 ng), Cy3-labeled telomeric probe (100 ng), sonicated male gDNA (3 μg) and sonicated salmon sperm DNA (25 μg).
Preparations for both FISH experiments were counterstained with 0.5 mg/mL DAPI, mounted in antifade based on DABCO (Sigma-Aldrich), and observed under a Zeiss Axioplan 2 microscope (Carl Zeiss Jena, Germany). Black-and-white images were recorded with a cooled monochrome CCD camera XM10 using cellSens Standard software, version 1.9 (Olympus Europa Holding, Hamburg, Germany). All images were captured separately for each fluorescent dye, pseudocolored (light blue for DAPI, green for fluorescein and red for Cy3) and superimposed with Adobe Photoshop, version 7.0.
Insect material and crossing strategy for the linkage map
Parents were derived from C. fumiferana 4th-6th instar larvae collected in 2013 from two Canadian populations in Alberta (AB; north of Conklin, 55.6985 N, -111.0841 W) and Quebec (QC; Gaspésie region, 48.4675 N, -68.1946 W). These two populations were selected on the basis of the large distance separating them (∼3000 km), each being near the longitudinal extremes of this species’ range in North America (Figure 1). We reasoned that genetic differentiation would likely be maximal between these two populations, thereby maximizing the number of informative loci between parents. Larvae were shipped to the Laurentian Forestry Centre (Quebec City, Canada) and reared to the adult stage. To obtain F1 families, we made 67 crosses according to the following combinations: QC male x QC female crosses (n = 20), QC male x AB female (n = 27), AB male x QC female (n = 12) and AB male x AB female (n = 8). Of these crosses, 39 were successful in producing progeny. This F1 generation was reared to the adult stage using rearing conditions favoring the bypassing of diapause (i.e., exposure to continuous light); in nature, 2nd instar spruce budworm larvae undergo winter diapause (i.e., dormancy) for about eight months. These rearing conditions allowed us to expedite the process of obtaining linkage-map families. Then, 86 backcrosses were made to obtain the F2 generation (see details in File S1). For backcross progeny, we used standard rearing conditions (i.e., with diapause) to achieve the highest rearing success possible. We successfully generated 24 F2 families, but most of them had fewer than 10 descendants. Given that substantial numbers of descendants tend to maximize the number of detectable recombination events used to build an informative linkage map, we selected the four largest unrelated F2 families available, representing 109 descendants in total (Figure 1). In addition, this multifamily mapping approach increased the probability of capturing segregating markers.
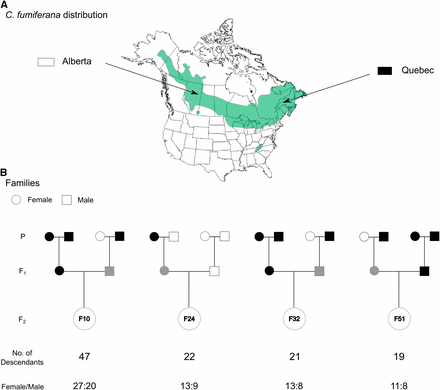
Sampling and crossing strategies employed to generate the Choristoneura fumiferana linkage map. (a) Map showing the geographic range of C. fumiferana in North America and the geographic origins of the two populations (Alberta and Quebec) used to generate the four families depicted below (adapted from Lumley and Sperling 2011). (b) Graphical representation of the family design used to generate the linkage map, with information on the number of descendants/family and the sex-ratio within each family. P, F1 and F2 labels identify, respectively, parent, first and second generations.
DNA extraction and sequencing
Before DNA extraction, thoraces and legs of adult moths were frozen in liquid nitrogen and ground using a Retsch MM 200 mixer mill (Retsch technology, Haan, Germany). Then, DNA was extracted with the DNeasy 96 Blood & Tissue Kit (Qiagen, Carlsbad, CA, USA) following the manufacturer’s instructions, with the exception of an additional RNase A treatment before the addition of buffer AL/ethanol (4 µL of 100 mg/mL Rnase A; 5 min digestion at room temperature). DNA purity of the extracts was assessed using a NanoDrop 8000 spectrophotometer (Thermo scientific, Waltham, MA, USA), and DNA concentration was evaluated using Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA). Samples were diluted to 20 ng/μL prior to library construction.
To prepare a reduced-representation library for sequencing, we used a modified genotyping-by-sequencing (GBS) protocol in which two restriction enzymes (PstI/MspI) and a Y-adapter were employed (Mascher et al. 2013). To ensure sufficient read depth, the library was sequenced on three P1v3 chips using HiQ reagents on an Ion Proton sequencer (Thermo scientific, Waltham, MA, USA). GBS library construction and sequencing were carried out at the Plate-forme d’analyses génomiques of the Institut de Biologie Intégrative et des Systèmes (IBIS) at Université Laval (Quebec City, QC, Canada).
SNP identification
Prior to analyses, read quality was assessed using the FastQC v. 0.11.1 software (Andrews 2016). SNP calling was carried out using both a reference-genome-independent method, i.e., the de novo Universal Network Enabled Analysis Kit (UNEAK) pipeline (Lu et al. 2013), and the Fast-GBS pipeline, which maps reads to a reference genome (Torkamaneh et al. 2017). This dual SNP calling approach was chosen to minimize the loss of genetic variation due to missing regions in the reference genome (Lu et al. 2013). Indeed, at the time these analyses were conducted, our draft C. fumiferana genome (bw6 version, Cusson et al. unpublished data) was thought to have missing regions that may be accessible using the uneak pipeline. uneak was run with the following parameter settings: minimum tag count c = 5 (default), error tolerance rate in the network filter e = 0.03 (default), and minimum minor allele frequency mnMAF = 0.01. Briefly, uneak retains all reads containing a barcode and a restriction enzyme cut site, in addition to being devoid of missing data in the first 64 bp after the barcode. Reads are then clustered into tags (i.e., reads displaying 100% identity), and only tags with c ≥ 5 are retained. Then, networks of tags differing by one bp are built. In these different networks, tags with read counts corresponding to 3% (e = 0.03) or less of read counts from the adjacent tags are considered errors. The edges connecting the “error” tags to “real” tags are then sheared, thus dividing networks into sub-networks or decreasing the number of tags present in networks. At the end of the process, only tag-pair networks are retained as potential SNPs; networks with multiple tags are discarded.
The procedure used by Fast-GBS for SNP identification first involves de-multiplexing and adapter trimming, keeping only reads ≥ 50 bp (user-defined). These reads are then mapped onto the reference genome, a step performed by the Burrows-Wheeler Aligner software package (BWA; Li and Durbin 2009) using the Maximal Exact match (MEM) algorithm. SNP calling is then carried out by Platypus v0.8.1 (Rimmer et al. 2014), using the following parameters: minimum number of supporting reads for a variant to be called (minReads) = 2, minimum read mapping quality (minMapQual) = 10, and minimum base-calling quality (minBaseQual) = 10.
SNP filtering
Missing data and genotyping errors hinder the correct ordering of markers and cause expansion of the linkage map (Hackett and Broadfoot 2003; Cartwright et al. 2007). To avoid these biases, we discarded SNPs genotyped in < 80% of individuals, as well as those showing a minimum allele frequency < 0.01 and those displaying heterozygosity > 0.75. In addition, we filtered out SNPs showing significant deviation from Mendelian or sex chromosome (Z-linked) inheritance patterns (P < 0.01) in one family out of four (see Chen et al. 2014). The first SNP filtering steps were carried out using VCFtools (Danecek et al. 2011), whereas the Mendelian/sex chromosome inheritance filtering was conducted using an in-house R script.
Linkage map construction
The linkage map was constructed using the Lep-MAP software (original version, Rastas et al. 2013). This software simultaneously analyzes thousands of markers from several outbred families and takes into account the absence of recombination in lepidopteran females (Robinson 1971; Suomalainen et al. 1973; Turner and Sheppard 1975). First, we employed the module EstimateLODLimit to choose the logarithm-of-odds (LOD) score threshold, which is used to determine whether or not markers belong to the same linkage group (LG). Based on 100 datasets randomly generated from the input data, an empirical distribution of maximum LOD scores is calculated from which the LOD value can be chosen with a desired significance level. Estimation of the LOD limit was made considering only maternal information (maleprior = -1). Then, assignment to LGs was carried out using the SeparateChromosomes module, on the basis of an LOD score limit of 6.8 (p value = 0.01) and considering only maternal information (maleprior = -1). At this step, only LGs with five or more markers were kept (sizeLimit = 5). Then, singular markers were added to the defined LGs, using the JoinSingles module with all informative markers and an LOD score limit of 8.4 (obtained by EstimateLODLimit with 7% significance and all informative markers). After the JoinSingles step, markers with more than 15 missing genotypes (13%) were removed from the LGs. Finally, ordering and positioning of markers was computed using the OrderMarkers module of Lep-MAP, with constant rates for genotype errors and recombination. For each LG, ten independent runs were carried out, at the end of which the marker ordering with the best likelihood was kept. LG-assigned markers with a genotype error rate estimate > 0.1 were then removed (genotype error rate estimation is given for each marker in the output of the OrderMarkers module). In addition, markers near the ends of LGs were also removed if they contributed to >1% of map length.
Synteny with the Bombyx mori genome
Reads containing SNPs used in generating the linkage map were mapped onto the genome of the silkworm, Bombyx mori (KAIKObase v. 3.2.2, http://sgp.dna.affrc.go.jp/KAIKO), using tblastx analysis (e-score cut-off value of 1.0e-03). KAIKObase has the advantage of providing chromosome assignments for the scaffolds forming the genome assembly of B. mori. As a complement to the above analysis, we also conducted a tblastx analysis against the genome of the Glanville fritillary, Melitaea cinxia (Lepbase version, http://lepbase.org), which has n = 31 chromosomes; this number corresponds to what is considered to be the ancestral number of chromosomes for the Lepidoptera (Suomalainen 1969; Lukhtanov 2000; Ahola et al. 2014; Yasukochi et al. 2016), thus making M. cinxia a valuable subject for syntenic analysis. Scaffold chromosome assignment for M. cinxia was obtained from the website of the Glanville fritillary genome project (Metapopulation research center, 2017, https://www.helsinki.fi/en/beta/metapopulation-research-centre/downloads; last consulted July 3, 2018). A graphical representation of the results of our C. fumiferana–B. mori synteny analysis was generated using the R package circlize (Gu et al. 2014).
Data availability
File S1 details the experimental design used to generate spruce budworm families for the linkage map. File S2 summarizes linkage map information i.e., number of markers and size in cM, for each linkage group. File S2 also provides details on position, errors and sequence for each marker included in the linkage map, and the significant hits in tblastx searches against the Bombyx mori and the Melitaea cinxia genomes. File S3 details assignment of the Choristoneura fumiferana linkage groups to B. mori and the M. cinxia chromosomes. Supplemental material and SNP data are available at Figshare: https://doi.org/10.25387/g3.6686543.
Results
Karyotype analysis
The molecular cytogenetic analyses of mitotic chromosomes of Choristoneura fumiferana confirmed the presence of 2n = 60 chromosomes in both males and females (Figure 2), i.e., the haploid chromosome number is n = 30. In both sexes, two chromosomes stood out by their larger size (Figure 2); in female preparations, one of these large chromosomes was more intensely stained with DAPI (blue) (Figure 2a; Figure 3e) and homogeneously highlighted with the green-labeled female gDNA probe (Figure 3d, f). These two large chromosomes represent the ZZ pair of sex chromosomes in males, and the WZ pair in females, where the intensely stained chromosome is the W chromosome composed of heterochromatin. Interestingly, in preparations of pachytene oocyte chromosomes stained by GISH and tel-FISH, the Z chromosome was clearly observed to wrap around the W chromosome to adjust its length for correct pairing; as a consequence, it appeared larger than its W counterpart (Figure 3d, e – see schematic representation). With GISH, strong binding of the female-derived genomic probe over the entire length of the W chromosome showed a high degree of molecular differentiation between the W and Z sex chromosomes (Figure 3d, f). The GISH analysis also highlighted a heterochromatic double-dot in one autosome bivalent in both sexes (Figure 3a-c; not shown for females) and an interstitial heterochromatic region of another autosome bivalent associated with the nucleolus (Figure 3d-f). This region obviously represents the nucleolus organizer region (NOR).
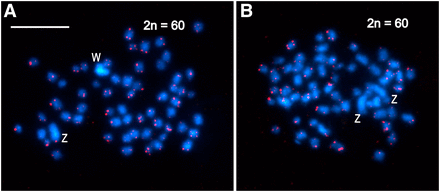
Karyotype analysis of mitotic chromosomes of Choristoneura fumiferana female (a) and male (b) by FISH using the (TTAGG)n telomeric probe. Hybridization signals of the Cy3-dUTP-labeled telomeric probe (red) indicate chromosome ends; chromosomes were counterstained with DAPI (blue). W and Z labels identify the largest chromosomes in mitotic complements, i.e., the sex chromosomes. (a) Mitotic metaphase of C. fumiferana female with a heterochromatic W chromosome and a large Z chromosome (2n = 60). (b) Mitotic metaphase of C. fumiferana male with two large Z chromosomes (2n = 60). Scale bar = 10 μm.
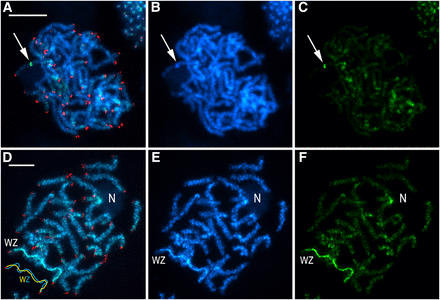
Genomic in situ hybridization combined with the (TTAGG)n telomeric probe in pachytene chromosomes in male (a-c) and female (d-f) Choristoneura fumiferana. Female-derived genomic probe was labeled with fluorescein-12-dUTP (green), and the telomeric probe with Cy3-dUTP (red); chromosomes were counterstained with DAPI (blue). Panels (a-c) show a male pachytene complement; arrows indicate heterochromatic block highlighted with the female genomic probe. Panels (d-f) show a female pachytene complement; “WZ” label identifies the sex chromosome bivalent (see schematic drawing in the lower left corner of panel d), where discrimination of the W chromosome is provided by the female-derived genomic probe; “N” indicates a nucleolus associated with a heterochromatic region (showing strong hybridization signals of the female genomic probe in panel d-f) of an autosome bivalent. (a, d) Merged images of preparations hybridized with female-derived genomic probe and telomeric probe, and counterstained with DAPI; (b, e) DAPI staining pattern; (c, f) hybridization pattern obtained using female-derived genomic probe. Scale bars = 10 μm.
SNP identification and filtering
The de novo UNEAK pipeline identified 208,098 polymorphic SNPs (Table 1), 1958 of which remained after eliminating those that did not genotype in at least 80% of individuals or did not present a minor allele frequency > 0.01. We then discarded an additional 679 SNPs that showed significant deviation from Mendelian or sex chromosome (Z-linked) inheritance patterns. In comparison, the reference-genome-based Fast-GBS pipeline generated 30,854 SNPs (SNPs having passed quality criteria in Platypus v0.8.1). Among these, we discarded markers that were not genotyped in at least 80% of individuals, presented a minor allele frequency < 0.01 or were triallelic. Of the remaining 22,094 SNPs, we discarded 9473 that showed significant deviation from Mendelian or sex chromosome (Z-linked) inheritance patterns. One descendant of family 24 (Figure 1) revealed Z-linked SNP segregation incompatible with the sex determined at the pupal stage; for the further analyses this individual was discarded. At the end of this filtering process, we were left with 13,900 SNPs, 12,621 and 1279 of which were identified using the Fast-GBS and UNEAK pipelines, respectively (Table1).
Number of SNPs retained after each filtering step as a function of the SNP identification pipeline used
| SNP filtering step | UNEAK | Fast-GBS | ||
|---|---|---|---|---|
| Filtered SNPs | Remaining SNPs | Filtered SNPs | Remaining SNPs | |
| Pipeline output | 208,098 | 30,854 | ||
| Genotypes in > 80% of individuals, minor allele frequency > 0.01 and diallelic1 | 206,140 | 1958 | 8760 | 22,094 |
| Deviation from mendelian or sex chromosome (Z-linked) segregation | 679 | 1279 | 9473 | 12,621 |
| Total SNPs used for the linkage map | 13,900 | |||
| SNP filtering step | UNEAK | Fast-GBS | ||
|---|---|---|---|---|
| Filtered SNPs | Remaining SNPs | Filtered SNPs | Remaining SNPs | |
| Pipeline output | 208,098 | 30,854 | ||
| Genotypes in > 80% of individuals, minor allele frequency > 0.01 and diallelic1 | 206,140 | 1958 | 8760 | 22,094 |
| Deviation from mendelian or sex chromosome (Z-linked) segregation | 679 | 1279 | 9473 | 12,621 |
| Total SNPs used for the linkage map | 13,900 | |||
The diallelic filtering step was run only for the Fast-GBS pipeline; the UNEAK pipeline automatically discards non-diallelic SNPs.
| SNP filtering step | UNEAK | Fast-GBS | ||
|---|---|---|---|---|
| Filtered SNPs | Remaining SNPs | Filtered SNPs | Remaining SNPs | |
| Pipeline output | 208,098 | 30,854 | ||
| Genotypes in > 80% of individuals, minor allele frequency > 0.01 and diallelic1 | 206,140 | 1958 | 8760 | 22,094 |
| Deviation from mendelian or sex chromosome (Z-linked) segregation | 679 | 1279 | 9473 | 12,621 |
| Total SNPs used for the linkage map | 13,900 | |||
| SNP filtering step | UNEAK | Fast-GBS | ||
|---|---|---|---|---|
| Filtered SNPs | Remaining SNPs | Filtered SNPs | Remaining SNPs | |
| Pipeline output | 208,098 | 30,854 | ||
| Genotypes in > 80% of individuals, minor allele frequency > 0.01 and diallelic1 | 206,140 | 1958 | 8760 | 22,094 |
| Deviation from mendelian or sex chromosome (Z-linked) segregation | 679 | 1279 | 9473 | 12,621 |
| Total SNPs used for the linkage map | 13,900 | |||
The diallelic filtering step was run only for the Fast-GBS pipeline; the UNEAK pipeline automatically discards non-diallelic SNPs.
Construction of a Choristoneura fumiferana linkage map
We ran Lep-MAP on 13,900 SNPs, 7610 and 8012 of which were maternally and paternally informative, respectively (i.e., heterozygous in the mother and the father of one of the crosses). The SeparateChromosomes module initially allocated 3180 SNPs to 30 LGs, while the JoinSingles module added 1853 SNPs to these 30 LGs, for a total of 5192 SNPs. After these two steps, 99 markers were removed from the LGs because more than 15 individuals presented missing genotypes (13%) for these markers. Then, the OrderMarkers module successfully ordered 3741 SNPs across 30 LGs. Among these 3741 SNPs, we discarded 50 markers presenting a genotype error estimate > 0.1 and 14 markers located near LG tips and contributing over 20 cM (>1.2%) to map length. In addition, we found 78 duplicated SNPs on the linkage map, a situation arising from our dual SNP identification approach (i.e., UNEAK + Fast-GBS pipelines). In these cases, we retained the SNP copy presenting the least amount of missing data, typically the one identified using the Fast-GBS pipeline. Finally, close examination of the SNPs identified using the UNEAK pipeline indicated that 13 of them were in fact duplicates that escaped detection due to the presence of an indel within a mono-nucleotide repetitive region of both alleles of a locus (for details on this property of UNEAK, see Picq et al. 2018). Once again, the SNP copy presenting the least amount of missing data were retained.
In the end, the GBS/SNP-based linkage map we developed for C. fumiferana is made up of 3586 markers forming 30 linkage groups (LGs), for a total length of 1720.41 cM (Figure 4, File S2). The number of LGs found is in agreement with the number of chromosomes identified by our cytogenetic analyses. The genetic length of each LG ranges from 24.72 to 110.22 cM, with an average inter-locus distance varying between 0.31 and 1.23 cM among LGs, and a number of SNPs ranging from 22 to 352 per LG. The longest LG, with 352 SNPs along 110.22 cM, revealed a SNP segregation pattern characteristic of the Z chromosome (female offspring appear homozygous for one of the father’s alleles, indicating hemizygosity). LG numbering is that provided by the Lep-Map software. Among the 3586 markers used to generate the linkage map, 3394 (94.65%) were identified using the reference-genome-dependent Fast-GBS pipeline, while the 192 remaining SNPs (5.35%) were found using the de novo UNEAK pipeline; 35 of these SNPs were in regions absent from our reference genome, confirming an earlier assessment of the bw6 assembly’s completeness. Thus, combining the two SNP identification approaches used here proved to be both appropriate and efficient, as judged by the quality and density of markers found, making it possible to obtain the expected number of linkage groups.

Linkage map generated for the spruce budworm, Choristoneura fumiferana, using the Lep-MAP software; based on 3586 SNP markers (vertical lines in each box). Linkage group numbering is derived from the Lep-MAP output.
It must be pointed out that the SeparateChromosomes module, parameterized with an LOD score limit of 6.8, identified a 31st LG featuring 39 SNP markers. However, the ordermarker module could order only 9 of these markers. After removing three markers that displayed a genotype error estimate > 0.1, we ran again the ordermarker module on the remaining markers of the 31st LG; this time, the ordermarker module was unable to order markers (blank output). To further explore this phenomenon, we ran the SeparateChromosomes module again, with a lower LOD score limit of 6.2. This time, the 39 SNP markers were added to the end of the 27th LG. After a first run of the ordermarker module followed by the deletion of markers displaying a genotype error estimate > 0.1, the ordermarker module was run a second time on this elongated LG. In the end, only 6 markers out of 39 could be successfully ordered, but they were 40 cM away from the nearest marker on LG 27 and spanned 91.41 cM. With these 6 additional markers, the LG 27 doubles in size. Eventually, the sequences associated with these 6 ordered markers did not generate significant hits against the B. mori genome or against the NCBI nr database (TBLASTX analyses, expected value cut-off: 1.0e- 03). In light of these results, we discarded the 31st LG obtained with the SeparateChromosomes module, parameterized with an LOD score limit of 6.8.
Chromosomal homology through synteny analysis
Among the sequences associated with the 3586 markers making up the C. fumiferana linkage map, 285 (7.9%) generated a significant hit in TBLASTX searches against the B. mori genome (expected value cut-off: 1.0e-03; Figure 5, File S2 and File S3). We inferred chromosomal homology between C. fumiferana and B. mori on the basis of 2 to 20 significant hits per chromosome. This analysis revealed one interesting feature: the Z chromosome of C. fumiferana corresponds to the fusion of chromosomes Z and 15 in the B. mori genome (Figure 5).
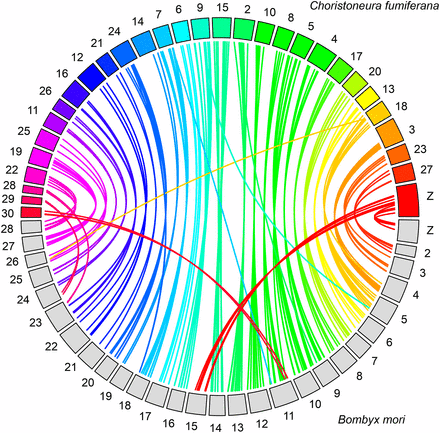
Mapping of Choristoneura fumiferana linkage groups onto Bombyx mori chromosomes. Each colored box represents one of the 30 C. fumiferana linkage groups (numbering from Lep-MAP output), while each gray box represents one of the 28 B. mori chromosomes (B. mori chromosome numbering; http://sgp.dna.affrc.go.jp/KAIKO). Each of the 285 connector lines identifies a one-to-one orthologous match between C. fumiferana and B. mori (TBLASTX analysis, expect value cut-off: 1.0e-03). Note that the C. fumiferana Z chromosome displays matches to both chromosomes Z and 15 in B. mori.
A similar synteny analysis targeting the M. cinxia genome yielded 176 significant TBLASTX hits and, overall, confirmed the above inferences about chromosome homology based on the B. mori genome (File S2 and File S3). However, LG 30 of C. fumiferana generated no significant hits in TBLASTX searches against M. cinxia chromosomes; in this case, chromosome homology was first inferred, and then confirmed with an earlier M. cinxia–B. mori synteny analysis (Ahola et al. 2014; see File S3 for details).
Given that the fusion of the Z chromosome with an autosome can significantly increase the adaptive potential of a species (Orr 2010), we conducted TBLASTX analyses against the NCBI nr database using, as queries, C. fumiferana sequences associated with markers located in the Z chromosome region corresponding to the B. mori chromosome 15 (Table 2). One Z-linked marker sequence revealed a significant match (expect value 5*10−6) with an ABC transporter C2 (ABCC2) protein of B. mori, which confers resistance to Bacillus thuringiensis toxin Cry1Ab (Atsumi et al. 2012).
SNP markers located in the Z chromosome region corresponding to B. mori chromosome 15 and identified in sequences displaying significant TBLASTX hits for known proteins
| Gene product | Function | Protein access. No. (organism) | No. of hits | Marker Identifier1 | Position (cM) | E-value | Marker position on B. mori chromosome 15 |
|---|---|---|---|---|---|---|---|
| Cap-specific mRNA (nucleoside-2’-O-)- methyltransferase transcript variant X2 | Modifies 5′ end of mRNA | XM_011563421.1 (Plutella xylostella) | 1 | 12 | 21.3 | 7.92*10−7 | 980069-980122 |
| Spectrin alpha chain transcript variant 3 | Molecular scaffold protein; determination of cell shape | XM_012692410.1 (Bombyx mori) | 2 | 22 | 31.88 | 10*10−6 | 1657717-1657661 |
| 23 | 31.88 | 5.07*10−5 | 1657810-1657754 | ||||
| Neuropathy target esterase/Swiss cheese | Membrane lipid homeostasis | XM_011570849.1 (Plutella xylostella) | 2 | 44 | 35.16 | 1.82*10−5 | 2595540-2595484 |
| 62 | 45.7 | 1.82*10−5 | 2595507-2595451 | ||||
| AP-3 complex subunit delta-1 | Facilitates budding of vesicle from the Golgi membrane | XM_012692363.1 (Bombyx mori) | 2 | 94 | 55.7 | 3.28*10−5 | 9224907-9224963 |
| 104 | 56.64 | 3.43*10−5 | 9226413-9226357 | ||||
| DNA binding protein RFX5 | Regulation of MHCII molecule transcription | XP_021189473.1 (Papilio xuthus) | 1 | 106 | 56.64 | 5*10−6 | 9598701-9598757 |
| Glutamate-cysteine ligase catalytic subunit | Glutathione biosynthesis | XM_004924611.2 (Bombyx mori) | 1 | 110 | 58.55 | 2.7*10−6 | 11234605-11234546 |
| Cyclic nucleotide-gated cation channel | Sensory transduction (including pheromone perception) | XM_012689102.1 (Bombyx mori) | 2 | 111 | 58.55 | 2.93*10−7 | 10837125-10837066 |
| 112 | 58.55 | 3.65*10−6 | 10837143-10837087 | ||||
| ABC transporter family C protein (ABCC2) | Transmembrane transport/detoxification | XM_012692465.1 (Bombyx mori) | 1 | 198 | 62.31 | 5*10−6 | 11251268-11251315 |
| Serine/threonine-protein phosphatase 2A | Removal of phosphate groups from phosphorylated Ser/Thr residues | XM_012697398 (Bombyx mori) | 1 | 203 | 62.31 | 2.5*10−5 | 13143820-13143764 |
| Gene product | Function | Protein access. No. (organism) | No. of hits | Marker Identifier1 | Position (cM) | E-value | Marker position on B. mori chromosome 15 |
|---|---|---|---|---|---|---|---|
| Cap-specific mRNA (nucleoside-2’-O-)- methyltransferase transcript variant X2 | Modifies 5′ end of mRNA | XM_011563421.1 (Plutella xylostella) | 1 | 12 | 21.3 | 7.92*10−7 | 980069-980122 |
| Spectrin alpha chain transcript variant 3 | Molecular scaffold protein; determination of cell shape | XM_012692410.1 (Bombyx mori) | 2 | 22 | 31.88 | 10*10−6 | 1657717-1657661 |
| 23 | 31.88 | 5.07*10−5 | 1657810-1657754 | ||||
| Neuropathy target esterase/Swiss cheese | Membrane lipid homeostasis | XM_011570849.1 (Plutella xylostella) | 2 | 44 | 35.16 | 1.82*10−5 | 2595540-2595484 |
| 62 | 45.7 | 1.82*10−5 | 2595507-2595451 | ||||
| AP-3 complex subunit delta-1 | Facilitates budding of vesicle from the Golgi membrane | XM_012692363.1 (Bombyx mori) | 2 | 94 | 55.7 | 3.28*10−5 | 9224907-9224963 |
| 104 | 56.64 | 3.43*10−5 | 9226413-9226357 | ||||
| DNA binding protein RFX5 | Regulation of MHCII molecule transcription | XP_021189473.1 (Papilio xuthus) | 1 | 106 | 56.64 | 5*10−6 | 9598701-9598757 |
| Glutamate-cysteine ligase catalytic subunit | Glutathione biosynthesis | XM_004924611.2 (Bombyx mori) | 1 | 110 | 58.55 | 2.7*10−6 | 11234605-11234546 |
| Cyclic nucleotide-gated cation channel | Sensory transduction (including pheromone perception) | XM_012689102.1 (Bombyx mori) | 2 | 111 | 58.55 | 2.93*10−7 | 10837125-10837066 |
| 112 | 58.55 | 3.65*10−6 | 10837143-10837087 | ||||
| ABC transporter family C protein (ABCC2) | Transmembrane transport/detoxification | XM_012692465.1 (Bombyx mori) | 1 | 198 | 62.31 | 5*10−6 | 11251268-11251315 |
| Serine/threonine-protein phosphatase 2A | Removal of phosphate groups from phosphorylated Ser/Thr residues | XM_012697398 (Bombyx mori) | 1 | 203 | 62.31 | 2.5*10−5 | 13143820-13143764 |
See File S2, linkage map details sheet, column named marker identifier.
| Gene product | Function | Protein access. No. (organism) | No. of hits | Marker Identifier1 | Position (cM) | E-value | Marker position on B. mori chromosome 15 |
|---|---|---|---|---|---|---|---|
| Cap-specific mRNA (nucleoside-2’-O-)- methyltransferase transcript variant X2 | Modifies 5′ end of mRNA | XM_011563421.1 (Plutella xylostella) | 1 | 12 | 21.3 | 7.92*10−7 | 980069-980122 |
| Spectrin alpha chain transcript variant 3 | Molecular scaffold protein; determination of cell shape | XM_012692410.1 (Bombyx mori) | 2 | 22 | 31.88 | 10*10−6 | 1657717-1657661 |
| 23 | 31.88 | 5.07*10−5 | 1657810-1657754 | ||||
| Neuropathy target esterase/Swiss cheese | Membrane lipid homeostasis | XM_011570849.1 (Plutella xylostella) | 2 | 44 | 35.16 | 1.82*10−5 | 2595540-2595484 |
| 62 | 45.7 | 1.82*10−5 | 2595507-2595451 | ||||
| AP-3 complex subunit delta-1 | Facilitates budding of vesicle from the Golgi membrane | XM_012692363.1 (Bombyx mori) | 2 | 94 | 55.7 | 3.28*10−5 | 9224907-9224963 |
| 104 | 56.64 | 3.43*10−5 | 9226413-9226357 | ||||
| DNA binding protein RFX5 | Regulation of MHCII molecule transcription | XP_021189473.1 (Papilio xuthus) | 1 | 106 | 56.64 | 5*10−6 | 9598701-9598757 |
| Glutamate-cysteine ligase catalytic subunit | Glutathione biosynthesis | XM_004924611.2 (Bombyx mori) | 1 | 110 | 58.55 | 2.7*10−6 | 11234605-11234546 |
| Cyclic nucleotide-gated cation channel | Sensory transduction (including pheromone perception) | XM_012689102.1 (Bombyx mori) | 2 | 111 | 58.55 | 2.93*10−7 | 10837125-10837066 |
| 112 | 58.55 | 3.65*10−6 | 10837143-10837087 | ||||
| ABC transporter family C protein (ABCC2) | Transmembrane transport/detoxification | XM_012692465.1 (Bombyx mori) | 1 | 198 | 62.31 | 5*10−6 | 11251268-11251315 |
| Serine/threonine-protein phosphatase 2A | Removal of phosphate groups from phosphorylated Ser/Thr residues | XM_012697398 (Bombyx mori) | 1 | 203 | 62.31 | 2.5*10−5 | 13143820-13143764 |
| Gene product | Function | Protein access. No. (organism) | No. of hits | Marker Identifier1 | Position (cM) | E-value | Marker position on B. mori chromosome 15 |
|---|---|---|---|---|---|---|---|
| Cap-specific mRNA (nucleoside-2’-O-)- methyltransferase transcript variant X2 | Modifies 5′ end of mRNA | XM_011563421.1 (Plutella xylostella) | 1 | 12 | 21.3 | 7.92*10−7 | 980069-980122 |
| Spectrin alpha chain transcript variant 3 | Molecular scaffold protein; determination of cell shape | XM_012692410.1 (Bombyx mori) | 2 | 22 | 31.88 | 10*10−6 | 1657717-1657661 |
| 23 | 31.88 | 5.07*10−5 | 1657810-1657754 | ||||
| Neuropathy target esterase/Swiss cheese | Membrane lipid homeostasis | XM_011570849.1 (Plutella xylostella) | 2 | 44 | 35.16 | 1.82*10−5 | 2595540-2595484 |
| 62 | 45.7 | 1.82*10−5 | 2595507-2595451 | ||||
| AP-3 complex subunit delta-1 | Facilitates budding of vesicle from the Golgi membrane | XM_012692363.1 (Bombyx mori) | 2 | 94 | 55.7 | 3.28*10−5 | 9224907-9224963 |
| 104 | 56.64 | 3.43*10−5 | 9226413-9226357 | ||||
| DNA binding protein RFX5 | Regulation of MHCII molecule transcription | XP_021189473.1 (Papilio xuthus) | 1 | 106 | 56.64 | 5*10−6 | 9598701-9598757 |
| Glutamate-cysteine ligase catalytic subunit | Glutathione biosynthesis | XM_004924611.2 (Bombyx mori) | 1 | 110 | 58.55 | 2.7*10−6 | 11234605-11234546 |
| Cyclic nucleotide-gated cation channel | Sensory transduction (including pheromone perception) | XM_012689102.1 (Bombyx mori) | 2 | 111 | 58.55 | 2.93*10−7 | 10837125-10837066 |
| 112 | 58.55 | 3.65*10−6 | 10837143-10837087 | ||||
| ABC transporter family C protein (ABCC2) | Transmembrane transport/detoxification | XM_012692465.1 (Bombyx mori) | 1 | 198 | 62.31 | 5*10−6 | 11251268-11251315 |
| Serine/threonine-protein phosphatase 2A | Removal of phosphate groups from phosphorylated Ser/Thr residues | XM_012697398 (Bombyx mori) | 1 | 203 | 62.31 | 2.5*10−5 | 13143820-13143764 |
See File S2, linkage map details sheet, column named marker identifier.
Discussion
We have combined cytogenetic approaches and a high-density SNP-based linkage map to explore the organization of the spruce budworm genome. Our karyotype analyses confirmed the presence of 30 pairs of chromosomes in Choristoneura fumiferana, a number commonly found in Tortricinae, one of three subfamilies recognized in the Tortricidae (Robinson 1971; Ennis 1976; Harvey 1997; Šíchová et al. 2013). Our molecular cytogenetic analyses also enabled identification of a noticeably larger pair of chromosomes in both sexes. In females, one of these two large chromosomes was strongly stained by both DAPI and fluorescently-labeled female gDNA (GISH analysis). In Lepidoptera, this staining pattern is typical of the W chromosome (Traut and Marec 1997; Mediouni et al. 2004; Fuková et al. 2005; Yoshido et al. 2006). As such, our findings provide evidence for the existence of a WZ/ZZ (female/male) sex chromosome system in the spruce budworm; C. fumiferana can now be added to the modest list of ∼90 lepidopteran species for which the sex chromosome system has been identified [F. Marec, unpublished data; Traut et al. (2007) and Marec et al. (2010) reported ∼40 species]. Interestingly, the number of lepidopteran species for which the sex chromosome system has been described is much smaller than the number for which chromosomes have been quantified (∼1,000; Robinson 1971; Ennis 1976; Blackmon et al. 2017). This difference may be attributed to the fact that many earlier studies used male germ cells, which are appropriate for chromosome counts, but not for identifying the sex chromosomes (Traut et al. 2007; Sahara et al. 2012). In recent years, the use of female meiotic pachytene chromosomes combined with techniques of molecular cytogenetics (FISH, CGH, GISH, etc.), has facilitated the characterization of sex chromosomes. Thus, in coming years, we can anticipate an increase in the number of species for which the sex chromosome system has been characterized (Sahara et al. 2012).
The ancestral chromosome number in Lepidoptera is inferred to be n = 31 (Suomalainen 1969; Lukhtanov 2000; Ahola et al. 2014; Yasukochi et al. 2016). From this ancestral state, the number of chromosomes has evolved through chromosome fission/fusion events (De Prins and Saitoh 2003; Brown et al. 2004; Marec et al. 2010). In tortricids, the reduced number of chromosome pairs relative to the ancestral state (i.e., n = 28 in Olethreutinae and n = 30 in Tortricinae, instead of 31) and the presence of large sex chromosomes suggest a sex chromosome-autosome fusion event during the evolution of their karyotypes (Fuková et al. 2005; Šíchová et al. 2013). Indeed, a recent cytogenetic study confirmed that the Z chromosome in three different tortricid species (Cydia pomonella, Lobesia botrana, and Eupoecilia ambiguella) arose from the fusion of an ancestral Z chromosome and an autosome (Nguyen et al. 2013). In our study, synteny analysis comparing the 30 linkage groups of C. fumiferana with the 28 chromosomes comprising the B. mori genome provided strong evidence for the presence of the same neo-Z chromosome in the spruce budworm genome. A Z chromosome-autosome fusion can have a significant impact on the adaptive capacity of a species. Indeed, the hemizygous state of the Z-linked genes in the female Lepidoptera increases the efficiency of natural selection acting on recessive mutations (faster X/Z evolution; Charlesworth et al. 1987; Bachtrog et al. 2009). In tortricids, the Z chromosome-autosome fusion is considered to have contributed to the adaptive success and the important radiation of this taxon (Nguyen et al. 2013). Indeed, the part of the neo-Z chromosome derived from an autosome corresponds to chromosome 15 in B. mori, which is known to bear clusters of genes involved in detoxification of plant metabolites and genes conferring insecticide resistance (Nguyen et al. 2013), such as the ABC transporter C2 (ABCC2) gene associated with recessive resistance to the Bacillus thuringiensis (Bt) toxin (Atsumi et al. 2012; Park et al. 2014; Xiao et al. 2014). In our study, the sequence of a marker located on the Z linkage group showed significant similarity (expect value: 5*10−6) to the ABCC2 protein located on chromosome 15 in B. mori. Thus, Z-linkage of the ABCC2 gene in C. fumiferana provides a genomic context favorable to the selection and rapid spread of Bt resistance in spruce budworm populations. Since there is significant variation in Bt tolerance among spruce budworm populations (van Frankenhuyzen et al. 1995), our results stress the importance of making rational use of Bt, as it is the main insecticide used to suppress spruce budworm populations in Canadian provinces (van Frankenhuyzen 2013).
The W chromosome of C. fumiferana displayed distinctive features relative to other chromosomes, including the Z chromosome. Indeed, the staining patterns obtained with our FISH assays revealed a W chromosome that is thoroughly and homogeneously heterochromatinized, i.e., constituted of condensed chromatin. Studies examining the composition of the heterochromatinized W chromosome in other lepidopterans have pointed to a high content of repetitive sequences, transposable elements, degenerated protein-coding genes, and sequences of mitochondrial origins (Abe et al. 2005; Fuková et al. 2007; Vítková et al. 2007; Traut et al. 2013; Lammermann et al. 2016). Such an unusual composition raises questions about the involvement of this chromosome in sex determination in Lepidoptera. For instance, in Samia cynthia, the W chromosome does not seem to be involved in sex determination (Yoshido et al. 2016). However, in B. mori, a dominant female-determining factor (Fem) located on the W chromosome has been shown to promote femaleness (Fujii and Shimada 2007). Recently, this feminizing factor was determined to be a small RNA (Fem piRNA, Kiuchi et al. 2014). Considering the above mentioned examples, the sex determination mechanism in the spruce budworm has a high chance of being specific to this species. In C. fumiferana and other pest species, identification of the sex determination mechanism has relevance beyond questions of basic evolutionary biology, as this knowledge could enable development of alternative pest management approaches based on the sterile insect technique (SIT) (Whyard et al. 2015; Darrington et al. 2017). The SIT method is based on rearing and releasing a large number of sterile males into wild populations with the aim to decrease reproductive success (Marec et al. 2005). RNA interference (RNAi) targeting genes involved in female sex determination has been used successfully to eliminate females during the rearing process, while RNAi could also be used to silence genes involved in spermatogenesis and induce male sterility (Whyard et al. 2015). SIT could thus offer an interesting alternative to the insecticide Btk (see above) to manage spruce budworm populations.
Concerning the heterochromatinization patterns of the W chromosome, those observed here in the spruce budworm are similar to patterns reported for the tortricid Cydia pomonella (Fuková et al. 2005; Šíchová et al. 2013). However, heterochromatinization of the W chromosome is partial in two other tortricid species, Lobesia botrana and Eupoecilia ambiguella (Šíchová et al. 2013). According to Šíchová et al. (2013), this pattern suggests that the tortricid W chromosome originated from the fusion of an ancestral W chromosome with an autosome, most likely the homolog of chromosome 15 in B. mori, as shown for the Z chromosome (see above). Thus, the spruce budworm appears to have a neo-W chromosome whose complete heterochromatization may have resulted from more rapid molecular degeneration of its neo-part than that affecting the W chromosomes of L. botrana and E. ambiguella.
To our knowledge, linkage maps have been published for 10 different lepidopteran species, half of which belong to the Papilionoidea superfamily (Tobler et al. 2004; Wang and Porter 2004, Jiggins et al. 2005; Kapan et al. 2006; Van’t Hof et al. 2008; Beldade et al. 2009; Winter and Porter 2010; The Heliconius Genome Consortium 2012; Rastas et al. 2013; Ahola et al. 2014; Li et al. 2015; Davey et al. 2016; Van Belleghem et al. 2017; Rastas 2017). The remaining linkage maps were developed for species belonging to different and somewhat distantly related superfamilies: Bombycoidea (Miao et al. 2005; Yasukochi et al. 2006; Yamamoto et al. 2008), Pyraloidea (Dopman et al. 2004), Yponomeutoidea (Heckel et al. 1999; Baxter et al. 2011), Geometroidea (Van’t Hof et al. 2013) and Noctuoidea (Cheng et al. 2017). In the superfamily Tortricoidea, attempts were made earlier to develop a linkage map based on 46 backcross progeny of two closely related budworms, C. occidentalis biennis and C. occidentalis occidentalis (Brunet 2014), but a limited number of SNPs (438) could be ordered and the number of linkage groups was higher (n = 51) than expected for this species (n = 30). Differences in accuracy and completeness between the C. occidentalis and the C. fumiferana linkage maps could be partly due to differences in genetic distance between the two pairs of populations used. Indeed, a recent study suggests that the genetic distance between the C. occidentalis biennis and C. occidentalis occidentalis populations sampled by Brunet (2014) was clearly lower than that between the C. fumiferana populations used in the present study (see Brunet et al. 2017). However, given that C. fumiferana and C. occidentalis are two closely related species (Dupuis et al. 2017), we will now be examining the possibility of combining the two data sets to further increase marker density on our linkage map.
Acknowledgments
This research was supported by a grant to MC from the Genomics Research and Development Initiative (GRDI; Government of Canada). Cytogenetic analyses were funded through grant 17-13713S from the Czech Science Foundation to FM.
Footnotes
Supplemental material available at Figshare: https://doi.org/10.25387/g3.6686543.
Communicating editor: B. Andrews
Literature Cited
Brunet, B. T. M., 2014 Genomic analysis of hybridization between the spruce budworm species Choristoneura fumiferana, C. occidentalis, and C. biennis (Lepidoptera: Tortricidae). PhD thesis, University of Alberta, Edmonton, AB, Canada.
Ministère des forêts, de la Faune et des Parcs, 2017 Aires infestées par la tordeuse des bourgeons de l’épinette qu Québec en 2017 - Version 1.0: Gouvernement du Québec, Direction de la protection des forêts.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}