





Abstract
The yeast Saccharomyces cerevisiae has emerged as a superior model organism. Selection of distinct laboratory strains of S. cerevisiae with unique phenotypic properties, such as superior mating or sporulation efficiencies, has facilitated advancements in research. W303 is one such laboratory strain that is closely related to the first completely sequenced yeast strain, S288C. In this work, we provide a high-quality, annotated genome sequence for W303 for utilization in comparative analyses and genome-wide studies. Approximately 9500 variations exist between S288C and W303, affecting the protein sequences of ∼700 genes. A listing of the polymorphisms and divergent genes is provided for researchers interested in identifying the genetic basis for phenotypic differences between W303 and S288C. Several divergent functional gene families were identified, including flocculation and sporulation genes, likely representing selection for desirable laboratory phenotypes. Interestingly, remnants of ancestor wine strains were found on several chromosomes. Finally, as a test of the utility of the high-quality reference genome, variant mapping revealed more accurate identification of accumulated mutations in passaged mismatch repair-defective strains.
Saccharomycescerevisiae is a genetically tractable model organism that is used to study a multitude of biological and disease processes (Botstein et al. 1997). There are many examples of the utility of yeast in uncovering fundamental biological pathways important for human health. For example, the elucidation of the conservation between yeast and human DNA mismatch repair contributed to the discovery that mismatch repair dysfunction was the causative agent in a common hereditary cancer syndrome (Fishel et al. 1993; Strand et al. 1993; Clark et al. 1999).
As yeast emerged as an important model organism, many laboratory strains were selected to express important characteristics such as the ability to mate, sporulate, and be transformed with high efficiency. Additionally, when manipulating yeast, researchers chose progeny lacking certain phenotypes such as agar invasion, clumping, and rapid sedimentation (Louis 2016). For example, S288C, a widely used laboratory strain (Goffeau et al. 1996), possesses a nonsense mutation in the FLO8 gene, which prevents clumping and invasive growth into agar, thereby allowing cells to be fully suspended in solution (Liu et al. 1996). W303, a descendant of S288C, was selected to retain the desirable characteristics of S288C, to sporulate well, and to be transformed with high efficiency (R. Rothstein, personal communication).
Differences among laboratory strains have been well documented; for example, analyses of the proteomes of several laboratory strains reveal differentially expressed proteins across various laboratory strains (Rogowska-Wrzesinska et al. 2001). Additionally, certain alleles of the SWI-SNF global transcription activator complex contribute to slow growth in the W303 background, but are lethal in S288C (Cairns et al. 1998). Given these differences, an understanding of the precise variations at the nucleotide level between strains is an important step in elucidating the underlying causes of phenotypic differences.
Since its origin, W303 has been widely used for genetic analyses of DNA repair and other biological mechanisms (Thomas and Rothstein 1989). Many of these studies require a reference sequence for genome-wide or hybridization-based molecular analyses. A high-quality reference genome would greatly improve these analyses, as well as provide insight into the unknown aspects of the evolutionary history of the strain. For example, S288C, D311-3A, and D190-9C are known to have contributed genetic information to W303; however, other ancestors remain unknown (R. Rothstein, personal communication and Rogowska-Wrzesinska et al. 2001).
For many years, a high-quality, chromosome length, annotated genome has existed for S288C; however, until this work, a similar resource did not exist for W303. Early draft genome sequence analyses of W303 suggested that W303 and S288C strains differed in ∼9700 easily identified nucleotide positions; however, more complex differences remained uncharacterized (Lang et al. 2013). W303 has been sequenced multiple times and these sequences are available in publicly accessible databases (Table 1); however, these sequences were not assembled into chromosomes or annotated and therefore were not useful to a broad range of scientific researchers. In this work, we present a chromosomally organized, annotated, high-quality genome reference for the W303 laboratory strain, along with a listing of the differences with the S288C reference genome. The resources can be utilized for genome-wide studies and comparative analyses. The genome sequence presented here represents a foundation for further improvement and curation, similar to the updates of S288C since the first completely sequenced genome appeared in the early 1990s (Goffeau et al. 1996; Engel et al. 2014).
Publicly available W303 sequencing data
| Reference | Platform | Coverage | Accession Number |
|---|---|---|---|
| Ralser et al. (2012) | Illumina and Roche-454 | 376× | GB: ALAV01000000 |
| Song et al. (2015) | Illumina | 301× | GB: JRIU01000000 |
| Lang et al. (2013) | Illumina | 300× | SRA: SRX315098 |
| Goodwin et al. (2015) | Oxford nanopore | GB: JSAC01000000 | |
| This work | PacBio | GB: LYZE00000000 |
| Reference | Platform | Coverage | Accession Number |
|---|---|---|---|
| Ralser et al. (2012) | Illumina and Roche-454 | 376× | GB: ALAV01000000 |
| Song et al. (2015) | Illumina | 301× | GB: JRIU01000000 |
| Lang et al. (2013) | Illumina | 300× | SRA: SRX315098 |
| Goodwin et al. (2015) | Oxford nanopore | GB: JSAC01000000 | |
| This work | PacBio | GB: LYZE00000000 |
GB, GenBank; SRA, NCBI Sequence Read Archive; PacBio, Pacific Biosciences.
| Reference | Platform | Coverage | Accession Number |
|---|---|---|---|
| Ralser et al. (2012) | Illumina and Roche-454 | 376× | GB: ALAV01000000 |
| Song et al. (2015) | Illumina | 301× | GB: JRIU01000000 |
| Lang et al. (2013) | Illumina | 300× | SRA: SRX315098 |
| Goodwin et al. (2015) | Oxford nanopore | GB: JSAC01000000 | |
| This work | PacBio | GB: LYZE00000000 |
| Reference | Platform | Coverage | Accession Number |
|---|---|---|---|
| Ralser et al. (2012) | Illumina and Roche-454 | 376× | GB: ALAV01000000 |
| Song et al. (2015) | Illumina | 301× | GB: JRIU01000000 |
| Lang et al. (2013) | Illumina | 300× | SRA: SRX315098 |
| Goodwin et al. (2015) | Oxford nanopore | GB: JSAC01000000 | |
| This work | PacBio | GB: LYZE00000000 |
GB, GenBank; SRA, NCBI Sequence Read Archive; PacBio, Pacific Biosciences.
Materials and Methods
Genomic DNA preparation and library construction
A 500 ml culture of wild-type W303 (MY7521) MATahis3-11,15 trp1-1, ura3-1 (derived from strains generously provided by Rodney Rothstein, Columbia University) was grown in rich medium for ∼24 hr. Genomic DNA extraction and purification was carried out according to Burke et al. (2000). Standard sequencing library construction was performed with Pacific Biosciences (PacBio) DNA Template Prep Kit 2.0 for one SMRT cell. The final library was sent to the University of California at Irvine for > 7 kb size selection shearing and sequencing with P6C4 chemistry.
Genome assembly and annotation
De novo assembly and polishing of PacBio reads was carried out with the Hierarchical Genome Assembly Process (HGAP) and QUIVER (Chin et al. 2013), resulting in 46× coverage. The 47 contig de novo assembly was scaffolded with datasets of shotgun sequences and unitigs of W303 (see Table 1) using the MeDuSa multi-draft scaffolding program (Bosi et al. 2015). Chromosome scaffolding was carried out with chromosome XII fragments in W303 and the corresponding BLAST hits to chromosome XII of S288C (NC_001144). Three unlocalized scaffolds representing the repetitive ribosomal DNA region on chromosome XII were removed before annotation (Venema and Tollervey 1999). To verify the scaffolding, sequencing reads of wild-type W303 (SRX315138) were mapped onto the draft assembly using the pipeline in Lang et al. (2013) with a quality threshold of 70. Regions without read coverage were considered scaffolding errors and were removed. Insertion/deletion (indel) error correction was conducted using high-quality Illumina wild-type W303 data (Lang et al. 2013, Table 1) with Pilon (Walker et al. 2014). Additionally, the completeness of scaffolds was determined by alignment of the de novo assembly and scaffolds to S288C version R64-2-1. Missing regions from chromosomes III and V were concatenated to corresponding scaffolds. Whole-genome and chromosome alignments were carried out with S288C using MAUVE (Darling et al. 2010) with match seed weight 15, full alignment, and iterative refinement.
The quality of the genome assembly was assessed with QUAST (Gurevich et al. 2013). Annotation was carried out with the Yeast Genome Annotation Pipeline (Proux-Wéra et al. 2012). Gene content between S288C and W303 was compared with OrthoVenn (Wang et al. 2015). Comparisons of sequence alignments and annotations were visualized with Geneious version 9.0.3 (Kearse et al. 2012).
Comparative analysis
MAUVE (Darling et al. 2004) whole-genome and chromosome alignments were used to analyze single nucleotide polymorphisms (SNPs) and rearrangements between W303 and S288C. MAUVE was utilized in order to identify the position of each polymorphic site in the reference and alternative genome sequence. MAUVE alignments and polymorphisms were visualized with genoPlotR (Guy et al. 2010) and Microsoft Excel.
Variants identified from the MAUVE (Darling et al. 2010) genome alignment of S288C and W303 were characterized with CooVar version 0.07 (Vergara et al. 2012) with respect to the position and coding sequences of S288C. MUSCLE (Edgar 2004; Li et al. 2015) alignments were analyzed to identify the conservation of repeat regions in Flo1 with the S288C ortholog of the protein (S288C: YAR050W). Divergent W303 orthologs (those with nonsynonymous substitutions) were analyzed with GO Slim Mapper (yeastgenome.org/cgi-bin/GO/goSlimMapper.pl) to determine whether variants mapped onto certain root biological processes. Genes that map onto the Saccharomyces Genome Database GO slim are listed in Supplemental Material, File S1. For analysis of sequence variations from S288C, megaBLAST (Zhang et al. 2000) alignments of each chromosome against the nucleotide collection were classified. The aligned sequences of the best hits (max score and E-value) for chromosomes III and XI were further analyzed due to similarity to chromosomes in strains distant from S288C. Whole-genome and chromosome phylogenies were calculated with CVTree3 for comparative analysis with K-tuple length 9. The genomes employed in the phylogenetic analysis are as follows: YJM1447 (GCA_000977955.1), YJM1388 (GCA_000977505.1), YJM1273 (GCA_000976995.1), YJM1248 (GCA_000976905.1), YJM681 (GCA_000976245.1), YJM244 (GCA_000975615.1) and EC1118 (GCA_000218975.1). Chromosomes III and XI of these assemblies were used for chromosome phylogenies.
Variant analysis of mismatch repair-deficient strains
Mapping of accumulated mutations in msh2 null (SRX315139) as well as msh2 missense variants—R542L (SRX315174), G688D (SRX315176), and A618V (SRX315175)—was carried out according to previous work (Lang et al. 2013) with more stringent quality filtering. Alignments with BWA (Li and Durbin 2009) mapping quality < 80 were ignored for variant detection purposes. Variants were detected using Freebayes (Garrison and Marth 2012) and filtered to include loci with depth of coverage > 10 and variant quality > 20, with the highest genotype quality of 5000.
Data availability
Strains are available upon request. The W303 sequences from this work are available at GenBank, accession number LYZE00000000. File S1 contains the characterized substitutions based on genome alignment of S288C and W303. File S2 contains the variant calling analysis with the improved W303 reference genome.
Results and Discussion
Alignment of S288C and W303 shows high similarity between the strains
The high-quality, chromosomally organized, annotated genome of the yeast strain W303 presented in this work was created by: (1) assembling long, lower fidelity reads (PacBio) into 47 contigs; (2) generating chromosome/episome length sequences using publicly available W303 data and S288C as scaffolds; and (3) error-correcting the assembled genome using short, high-fidelity reads. The complete W303 genome statistics are shown in Table 2. The genome is made up of 18 scaffolds that represent the 16 chromosomes, the mitochondrial genome, and the 2 μm plasmid.
W303 genome assembly statistics
| Reference (S288C) | Initial W303 Assembly | Current W303 Assembly | |
|---|---|---|---|
| Number of contigs/scaffolds | 17 | 47 | 18 |
| Largest contig/scaffold | 1,531,933 | 1,526,194 | 1,575,129 |
| Total length | 12,157,105 | 12,658,946 | 12,423,513 |
| GC (%) | 38.2 | 38.28 | 38.18 |
| N50a | 924,431 | 605,842 | 929,095 |
| Reference (S288C) | Initial W303 Assembly | Current W303 Assembly | |
|---|---|---|---|
| Number of contigs/scaffolds | 17 | 47 | 18 |
| Largest contig/scaffold | 1,531,933 | 1,526,194 | 1,575,129 |
| Total length | 12,157,105 | 12,658,946 | 12,423,513 |
| GC (%) | 38.2 | 38.28 | 38.18 |
| N50a | 924,431 | 605,842 | 929,095 |
N50 is the weighted median statistic such that 50% of the entire assembly is contained in contigs/scaffolds equal to or larger than this value. The initial assembly is after Hierarchical Genome Assembly Process pipeline assembly of raw reads. The current assembly has undergone scaffolding with MeDuSa and removal of scaffolding errors.
| Reference (S288C) | Initial W303 Assembly | Current W303 Assembly | |
|---|---|---|---|
| Number of contigs/scaffolds | 17 | 47 | 18 |
| Largest contig/scaffold | 1,531,933 | 1,526,194 | 1,575,129 |
| Total length | 12,157,105 | 12,658,946 | 12,423,513 |
| GC (%) | 38.2 | 38.28 | 38.18 |
| N50a | 924,431 | 605,842 | 929,095 |
| Reference (S288C) | Initial W303 Assembly | Current W303 Assembly | |
|---|---|---|---|
| Number of contigs/scaffolds | 17 | 47 | 18 |
| Largest contig/scaffold | 1,531,933 | 1,526,194 | 1,575,129 |
| Total length | 12,157,105 | 12,658,946 | 12,423,513 |
| GC (%) | 38.2 | 38.28 | 38.18 |
| N50a | 924,431 | 605,842 | 929,095 |
N50 is the weighted median statistic such that 50% of the entire assembly is contained in contigs/scaffolds equal to or larger than this value. The initial assembly is after Hierarchical Genome Assembly Process pipeline assembly of raw reads. The current assembly has undergone scaffolding with MeDuSa and removal of scaffolding errors.
To analyze the divergence of W303 from its parent strain, S288C, the genomes were aligned using MAUVE (Darling et al. 2010). Figure 1 shows the collinear blocks of homology between the strains. In Figure 1, each colored segment represents a distinct region of DNA that shares homology without gaps or rearrangement. Despite some telomeric rearrangements, S288C and W303 are highly similar in genomic structure and sequence identity. The alignment of chromosome IX exhibits high synteny with S288C and shows only one breakpoint between the collinear homologous regions of the chromosome (Figure 1).
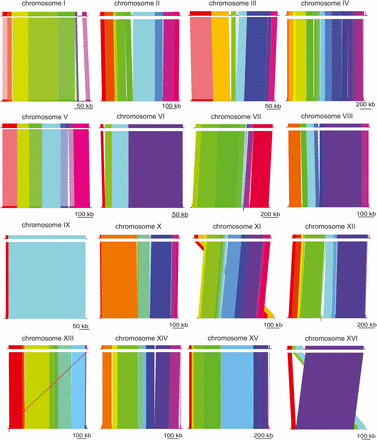
Highly similar genome structure between W303 and its ancestor, S288C. Chromosome alignments of W303 (top) and S288C (bottom) are shown. The color blocks do not signify the degree of sequence similarity, instead they represent stretches of homology without gaps or rearrangements. Scale bars are shown for reference below each alignment.
In contrast, chromosome XVI shows rearrangement near a terminal region of the chromosome. A transposable element and a Y’-encoded ATP helicase flank the junction of this region. This finding is not surprising because both transposable elements (Mieczkowski et al. 2006) and Y’-helicases, thought to have originated as mobile elements, are associated with chromosomal rearrangement and recombination (Louis and Haber 1992; Schmidt and Kolodner 2006).
The divergence in telomeric regions includes changes beyond the large rearrangement discussed above. A comparison of the gene content between S288C and the annotation of W303 shows expansion of Y’ element ATP-dependent helicase protein throughout the genome, including the acquisition of Y’ regions on chromosomes without these subtelomeric elements in S288C. These differences were identified on the right arm of chromosomes III and XIV (Louis et al. 1994; Louis 1995). Previous work demonstrated that subtelomeric elements undergo recombination and expansion in telomerase-deficient strains in order to restore telomeres (Lundblad and Blackburn 1993). and that the presence of these Y’ helicases varies between related strains of S. cerevisiae on homologous chromosomes (Chan and Tye 1983).
Although the chromosome structure (Figure 1) and lengths (Figure 2A) of W303 and S288C are similar, there are 9500 single nucleotide variations (Figure 2A and File S1). Figure 2B shows that at the nucleotide level, some chromosomes are more homologous to S288C (blue), while others are more divergent (gray). Overall, chromosome XI is the most distinct from its respective chromosome in S288C (Figure 2B). This observation prompted further analysis of the divergence or ancestry at the chromosome level.
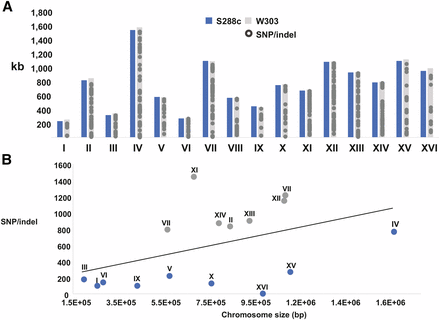
Sequence differences identified in W303 and S288C strains. (A) The chromosome sizes in kilobases of S288C (blue) and W303 (gray) are shown for comparison. The distribution of SNPs or small indels across the positions within the 16 chromosomes are shown (gray circles). In many regions, the density of polymorphisms is such that individual sites of change are not distinguishable. (B) The relationship between the number of SNPs or indels and the length of the chromosome in base pairs is shown. The chromosome number is displayed above the symbol. When comparing the differences per chromosome, two classes emerge: chromosomes that are more divergent from S288C (gray) or more similar to S288C (blue). Indels, insertions/deletions; SNP, single nucleotide polymorphisms.
While S288C and W303 are highly similar, each chromosome was analyzed to identify regions that may be divergent. After performing megablast BLASTn (Zhang et al. 2000) alignments against the nucleotide collection, W303 chromosomes III and XI were found to share significant sequence identity to the respective chromosomes in strains: EC1118, max score = 2.018e + 05, E value = 0.0 (Novo et al. 2009) and YJM244, max score = 2.461e + 05, E value = 0.0 (Strope et al. 2015). Interestingly, both are wine fermentation strains with European ancestry. These regions of similarity with the wine strains include continuous regions of chromosome III and XI. In contrast, when these same regions in W303 were aligned with S288C, the output showed shorter segments of homology with multiple gaps.
Phylogenetic analysis of several S. cerevisiae strains from various populations confirms the close relationship between S288C and W303 genome-wide (Figure 3A). Interestingly, S288C and W303 branch in a clade with the commercial wine strain EC1118 mentioned above (Figure 3A). This sequence similarity may reflect a shared wine strain ancestry among these three strains. To determine whether W303 has distinct wine strain ancestry, chromosome alignments of the S. cerevisiae strains described above were conducted to identify polymorphisms among the strains. The analysis revealed identical polymorphic sites shared among the wine strains EC1118 and YJM244, and the laboratory strains S288C and W303, on chromosomes III and XI (Figure 3B, COMMON). Interestingly, on W303 chromosome XI, which is the most divergent from S288C (Figure 2B), there are many polymorphic sites that are distinct from S288C, but identical to ones in the wine strains EC1118 and YJM244 (Figure 3B, DIVERGENT).
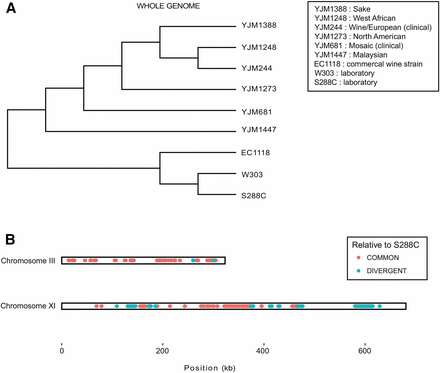
Phylogenetic analyses reveal potential remnants of wine strain ancestry. (A) Phylogeny of whole genomes of S. cerevisiae strains from various populations. A key of the populations associated with each strain are given in the upper right rectangle. (B) Identified polymorphisms across yeast species. The polymorphisms were identified using MAUVE chromosomal alignments with the strains shown in (A). Sites with common nucleotides only in the EC1118 and YJM244 wine strains, W303, and S288C are shown as points in orange (COMMON), while blue points represent potential sites of divergence from S288C where sites are only identical across the EC1118 and YJM244 wine strains and W303 (DIVERGENT).
W303 ancestors include D311-3A and D190-9C, strains with unknown ancestry (R. Rothstein, personal communication and Rogowska-Wrzesinska et al. 2001). The data presented in this paper suggest that these strains might also have European wine ancestry. Further sequencing of laboratory strains in the pedigree of W303 would allow for the characterization of the source of the divergence of W303 from S288C.
Divergent coding sequences of W303 compared to S288C
To analyze potential functional consequences of the differences between W303 and S288C, synonymous and as well as the conservative and nonconservative nonsynonymous substitutions were characterized using Coovar version 0.07 (Vergara et al. 2012). The analysis was based on the variants identified from MAUVE (Darling et al. 2010) genome alignment between S288C and W303. The results are provided as a comprehensive listing of the genomic variation between the strains that may be a useful tool for researchers interested in understanding the genetic basis of phenotypic differences (File S1).
Because nonsynonymous substitutions have the capacity to have biological consequences, the complete list of highly divergent genes with the number of conservative and nonconservative nonsynonymous substitutions is supplied in File S1. The variants with nonsynonymous changes were mapped to Gene Ontology (GO) terms. There was not a significant enrichment in any category for the entire group of ∼700 genes with nonsynonymous differences or with the ∼220 genes with nonconservative substitutions (File S1).
Although there was not enrichment in a specific functional category, certain genes were strikingly divergent; for example, YHL008C, an uncharacterized open reading frame, sustained substantially more nonsynonymous substitutions than the any other gene. YHL008C has 83 nonsynonymous substitutions (11 of which are nonconservative) over the 1884 nt open reading frame. Little is known about the function; however, deletion of this open reading frame decreases chloride accumulation (Jennings and Cui 2008). Early yeast transformation procedures often employed calcium chloride to increase transformation efficiency. As mentioned previously, W303 was selected to have superior transformation efficiency over S288C. Variations in YHL008C and the other 42 coding sequences involved in ion transport (GO:0006811, File S1) might be associated with the selection of spores with high transformation efficiency during crosses that gave rise to W303.
The second gene with the most nonconservative, nonsynonymous substitutions is AAD4, an aryl alcohol dehydrogenase (AAD). The W303 AAD4 gene has 48 variants (nine conservative and nine nonconservative substitutions) in the 990 nt open reading frame. Variability in AADs has been associated with wine and other fermentation strains (Borneman et al. 2011). The AAD enzymes convert aldehydes and ketones into their corresponding aromatic alcohols. As such, the variability of AAD genes in different fermentation yeast strains is thought to influence the volatile aromas produced during wine fermentation, and aroma characteristics are an important component of wine quality (Li et al. 2014).
With an understanding of the history of W303, we examined certain other processes that had been selected for during the crosses to create the strain. As mentioned above, W303 was selected to have a higher sporulation efficiency than S288C (Gerke et al. 2006) (R. Rothstein, personal communication). Interestingly, differences in 19 of 176 sporulation genes (GO:0043934) (Hong et al. 2008) were identified.
Similarly, selection against flocculation in ancestral laboratory strains likely gave rise to lessened selective pressure of these genes. As mentioned above, S288C harbors an inactivating point mutation in FLO8, whose gene product is a transcription factor required for flocculation and invasive growth (Liu et al. 1996). In W303, there are 13 aa differences in the 2277 nt open reading frame for Mss11, a protein that coregulates cell wall genes with Flo8 (Bester et al. 2012). Additionally, another flocculation gene, FLO9, harbors 12 nonsynonymous substitutions (two nonconservative) in an open reading frame of 3969 nt. Finally, the W303 FLO1 gene maintains expansions in the flocculin repeat region (Figure 4) that directly correlate with adhesion phenotypes (Verstrepen et al. 2005). SPSC01, a constitutively flocculent strain, contains an expansion of these domains in Flo1 (He et al. 2012). The variation in this region may be due to instability at these repetitive regions, or reflect a more flocculent ancestor of W303. Taken together, these divergences might be a consequence of changes that occur in the continuous laboratory selection against the flocculation function.
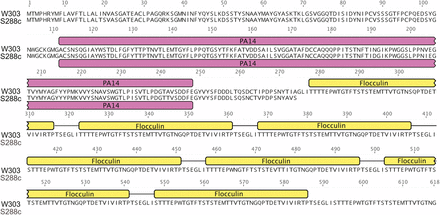
Divergent coding sequences in W303 when compared to S288C. Divergent regions of the protein sequence of Flo1 are highlighted in the alignment with other variants of the protein residues (S288C: YAR050W). Protein domains are shown above the alignment, PA14: pink, flocculin: yellow. Expansions of the flocculin repeats in W303 are shown.
Although only a few observations are cited above, the analysis of the polymorphisms identified from alignment of S288C and W303 should serve as a tool to begin to understand the mechanisms underlying phenotypic variations between the strains.
Improvement of mapping of mutation accumulation in a mismatch repair-defective strain
The assembled genome sequence of W303 described in this work was employed to validate the efficacy in accurate mutation calling. Previously, we conducted a mutation accumulation analysis with a lower quality S288C SNP-adjusted draft genome that required the manual verification of all called single base substitutions and indels at repetitive elements (Lang et al. 2013). By manual verification, we refer to final steps in the SNP calling pipeline to eliminate false positives. The process includes filtering out commonly called false positives and then visualizing the aligned sequencing reads of the passaged strains along with the ancestors using genome viewing software to verify the fixed mutations in the passaged mutator strains (Lang et al. 2013). In the previous analysis, the identification of insertion/deletion mutations required less stringent SNP calling parameters; however, while capturing the mutations, the less stringent SNP calling output resulted in a large number of false positives. We reasoned that high-throughput mapping of mutaions, particularly insertion/deletions, should be more accurate and require less manual verification with a higher quality reference genome. To test this, DNA reads from serially passaged mismatch repair-deficient strains (Lang et al. 2013) were mapped onto the S288C SNP-adjusted W303 draft genome (Lang et al. 2013) and to the high-quality W303 genome presented in this work. The SNP calling parameters were similar to those used previously with minor modifications (described in the Materials and Methods).
As anticipated, an improvement in the number of calls was observed with the high-quality W303 genome in contrast to the SNP-adjusted S288C draft genome (Figure 5). For example, with the null msh2 passaged strain, mapping onto the current high-quality W303 assembly decreased insertion or deletion calls from 422 to 248 and the number of SNP calls decreased from 138 to 44. The in all cases, the number of SNP variants called with the high-quality genome was closer to the actual number of mutations verified manually (Figure 5 and File S2). Importantly, the mutations identified in mismatch repair-defective strains using the high-quality W303 assembly recapitulate the increased identification of insertions and deletions, without creating a large number of false positives (Figure 5). In conclusion, these data represent an improvement on the S288C SNP-adjusted draft W303 genome and can be employed for analysis of the ancestry, variant detection, and other genome-wide studies.
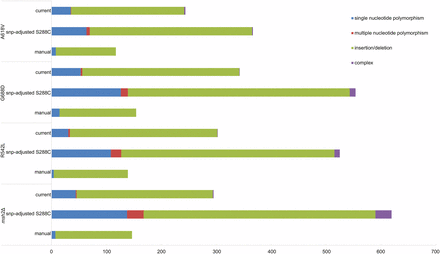
Mutation calling using the high-quality W303 genome is similar to manually verified mutation numbers. Mapping was employed to compare the variant identification between the SNP-adjusted S288C draft (Lang et al. 2013) and the current high-quality genome assembly. Purple, complex (consecutive indels and polymorphisms); green, indels, red; multiple nucleotide polymorphisms (consecutive SNPs); blue, SNP. Indels, insertions/deletions; SNP, single nucleotide polymorphisms.
Acknowledgments
The authors would like to acknowledge Wei Wang, Jessica Wiggins, and the Princeton Sequencing Core Facility for their support of this project. We would also like to acknowledge Mark Rose for his support of this work. The research that is the basis of this manuscript was conducted at Princeton University. This research was supported by a New Jersey Commission on Cancer Research Seed grant (10-1064-CCR-E0) and a Cancer Institute (NCI) of New Jersey NCI Cancer Center Support grant (P30 CA072720). K.M. was partially supported by a T32 GM007388 training grant. Some equipment and supplies were purchased through R01 GM037739. The Princeton Sequencing Core Facility was supported in part by the National Institute of General Medical Sciences (P50 GM071508). The authors declare that they have no competing interests.
Author contributions: K.M. and A.G. conceived the study. L.P. assembled the genome sequence. K.M. carried out all subsequent analysis. K.M. and A.G. wrote the manuscript. All three authors read and approved the final manuscript.
Footnotes
Supplemental material is available online at www.g3journal.org/lookup/suppl/doi:10.1534/g3.117.040022/-/DC1.
Communicating editor: B. J. Andrews
Literature Cited
Louis, E. J., 2016 Corrigendum: historical evolution of laboratory strains of Saccharomyces cerevisiae. Cold Spring Harb. Protoc.
Author notes
This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Present address: National Institute of General Medical Sciences, National Institutes of Health, Bethesda, MD 20892-6200.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}