





Abstract
We describe a suite of predictive models, coined FASTmC, for nonreference, cost-effective exploration and comparative analysis of context-specific DNA methylation levels. Accurate estimations of true DNA methylation levels can be obtained from as few as several thousand short-reads generated from whole-genome bisulfite sequencing. These models make high-resolution time course or developmental and large diversity studies practical regardless of species, genome size, and availability of a reference genome.
Advances in high-throughput sequencing has allowed for single-base resolution analysis of DNA methylation at cytosines across an entire genome. This was first applied to the model plant Arabidopsis thaliana (Cokus et al. 2008; Lister et al. 2008) and, since then, has been applied to numerous species, including protists, fungi, insects, anthozoa, tunicates, fish, and mammals (Lister et al. 2009; Feng et al. 2010; Zemach et al. 2010). Currently, DNA methylation is profiled genome-wide by deep, whole-genome bisulfite sequencing (WGBS). The use of a reference genome is essential to inform the methylation status at each cytosine reference position, where a thymine in lieu of cytosine indicates an unmethylated cystosine (Urich et al. 2015). Thus, absence of a reference genome has prevented rapid, genome-wide analysis of DNA methylation for the majority of known species, and is cost-prohibitive for high-resolution developmental or time-course studies in species with large genomes. To date, several methods exist to accommodate the challenges associated with non-reference-based analysis of DNA methylation, but they lack cytosine context sequence specificity (Kuo et al. 1980; Fraga et al. 2002; Karimi et al. 2006).
Here we present FASTmC, a suite of predictive models that can be used to estimate genome-wide DNA methylation levels at all cytosine sequence contexts without the use of a reference genome. These models assumed a relationship between DNA methylation levels calculated from alignment of WGBS reads to a reference genome (target; ) and from direct assessment from raw WGBS reads (i.e., no alignment to a reference genome) (estimator; ) (Supporting Information, Table S1). Methylation levels are calculated as the proportion of methylated cytosines to the total number of possible methylated cytosines. The difference between the two variables exists at unmethylated cytosines; the estimator value includes unmethylated cytosines and true thymines when calculating the DNA methylation level. Estimator DNA methylation levels were compared to target levels to determine a relationship, and the strength of which, to confidently predict/extrapolate genome-wide DNA methylation levels for any sample regardless of the availability of a reference genome.
Using publicly available data, for species with reference genomes, actual and estimator DNA methylation levels for 44 species were used to construct models capable of predicting genome-wide levels of DNA methylation for species without a sequenced genome. Using additional publicly available data from mutants and cell-types known to be different from wild-type samples, we discuss the sensitivity, robustness, and utility of the models in terms of CpG DNA methylation, followed by plant- (CHG and CHH) and mammal-specific (CH) DNA methylation.
Materials and Methods
WGBS data were downloaded from the Short Read Archive (SRA)/Gene Expression Omnibus (GEO) or sequenced in-house (Table S1). WGBS data were aligned using methods described in (Schultz et al. 2015) to generate “allC” files. The allC files were used to determine target DNA methylation levels, and can be downloaded from GEO under accession number GSE72155. Prior to estimation of predictor DNA methylation levels, WGBS data were trimmed of adaptor sequences using Cutadapt v1.9 (Martin 2011), end-trimmed using Trimmomatic (Bolger et al. 2014), and quality filtered using FASTX-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Reads of at least 30 bp in length with ≥ 20% of nucleotides having a quality score ≥ 75% were retained. Random sampling without replacement was performed with increasing fold-change from 1 to 105 reads using the program fastq-tools (http://homes.cs.washington.edu/~dcjones/fastq-tools/). Custom Perl scripts were used to sum the number of and sites for each randomly sampled read, and subsequently to estimate the predictor DNA methylation level at CpG, CHG, CHH, and CH sites (Table S1). A characteristic shared among all studies utilizing WGBS data is the inability to distinguish between 5-methylcytosine and 5-hydroxymethylcytosine (5hmC) (Huang et al. 2010). Therefore, levels of DNA methylation represent both forms of methylated cytosines, although it should be noted there is no evidence for 5hmC in plant genomes (Erdmann et al. 2014).
Violation of the assumptions can cause inaccuracies in estimating . We discuss some of these violations in Results and Discussion. In addition, we note that when additional genomic short read data (≥ 500,000 bp) are available, GC content and the frequency of the target site in the genome, e.g., the frequency of CpG dinucleotides, can be directly measured. This can then be used to directly calculate the proportion of target sites that are methylated, , using the frequency of intact target sites, e.g., CpG, that remain in the bisulfite genome data. These are sites that were methylated and thus escaped C to T conversion.
Data availability
All data used in this study can be found on the SRA/GEO webpages. Accession identifiers can be found in Table S1.
Results and Discussion
FASTmC is able to detect intraspecific differences in DNA methylation (Figure 1). In the plant A. thaliana, mutants exist that are defective for enzymes that are required for maintenance of CpG DNA methylation – met1, met1+cmt3, and vim1+vim2+vim3 – as they have reduced CpG methylation levels compared to wild-type (Stroud et al. 2013). Also, several mutant genotypes for met1 show different degrees of loss of CpG DNA methylation compared to each other: (i) an original met1 mutant genotype (high loss); (ii) a met1 heterozygous mutant genotype (met1+/−) (intermediate loss); and (iii) a recovered genotype (MET1+/+) from a MET1+/+ and met1+/− backcross. The recovered MET1+/+ is wild-type for MET1 function but has lost CpG methylation in some regions of the genome (low loss). FASTmC is able to capture the differences between these different genotypes (Figure 1A). Additionally, the slight (∼3%) difference between MET1+/+ and the met1+/− mutant can be distinguished, demonstrating the sensitivity of FASTmC (Figure 1A).
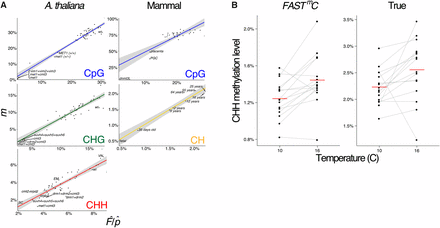
Detection of intraspecific DNA methylation levels by FASTmC. (A) Linear models (LMs) for estimated levels of methylation, i.e., , (Y-axis) vs. actual levels (X-axis) determined by reference mapping of WGBS reads. Estimated levels of methylation were based on 10,000 random WGBS reads. DNA methylation differences between A. thaliana mutants, mouse mutants/cell-types/tissues, and increasing CH methylation throughout brain development are captured with FASTmC. Shaded area represents the 95% confidence interval. (B) Environmental (temperature) effects on CHH DNA methylation in A. thaliana is also recapitulated using FASTmC. Left panel (“FASTmC”) represents FASTmC methylation estimates for individual lines based solely on the WGBS reads using the “plant” model from http://fastmc.genetics.uga.edu. Right panel (“True”) represents the methylation values from standard WGBS read alignment to the A. thaliana reference genome. Red lines are averages of all lines. Data from Dubin et al. 2015.
FASTmC is also capable of capturing natural epigenetic variation exhibited by changes in levels of CHH methylation due to temperature in A. thaliana (Figure 1B). Levels of CHH methylation are affected by temperature such that a higher level is observed at higher temperatures as opposed to lower temperature treatments (Dubin 2015). Applying FASTmC to these published data (Dubin 2015) using a fraction of the original WGBS data recapitulated these findings (Figure 1B). Thus, studies investigating natural epigenetic variation can be performed at a fraction of the cost.
In mammals, epigenetic reprogramming, including CpG demethylation, is required to erase DNA methylation imprints and epimutations established in the previous generation (Reik et al. 2001). Following demethylation, DNA methylation patterns are reestablished at imprinted loci and transposable elements (TEs) during gametogenesis by the de novo methyltransferases DNMT3A and a noncatalytic paralog, DNMT3-like (DNMT3L) (reviewed by Law and Jacobsen 2010). The reductions in CpG DNA methylation caused by epigenetic reprogramming in primordial germ cells (PGCs) or by mutations in DNMT3L (dnmt3L) compared to somatic tissues are captured by FASTmC (Figure 1A) (Popp et al. 2010; Kobayashi et al. 2012; Seisenberger et al. 2012). Additionally, increased levels of CpG DNA methylation in the brain (e.g., NeuN+ and glia cells) (Lister et al. 2013) can be differentiated from other somatic tissues (Figure 1A and Table S1) (Hon et al. 2013). Overall, as demonstrated in A. thaliana and Mus musculus, FASTmC can be used to detect intraspecific differences of DNA methylation levels at CpG sites (Figure 1A).
We determined natural interspecific variation of DNA methylation at CpG sites across 44 different species (Figure 2). However, unlike intraspecific comparisons between mutants or cell types, nucleotide biases, such as genomic GC content differences, can over- or underestimate the estimator value for the CpG sequence contexts. The estimator (see Equation 2) is estimating the product of the methylation frequency of CpG sites and the GC content of the genome, and is thus confounded. This bias can be overcome in all species investigated but mammals (Homo sapiens, M. musculus, and Canis lupus familiaris) by dividing the estimator value by an average GC content of the genome, which corrects the relationship between target and estimator to ∼1:1. GC content can be approximately estimated from WGBS reads (see Materials and Methods) or additional genomic sequence data – 10,000 50-bp reads (500,000 bp) – can be used to directly estimate GC content (Table S1).
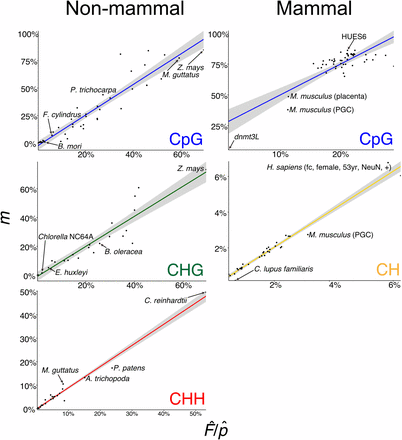
Detection of interspecific DNA methylation levels by FASTmC. Linear models (LMs) for estimator () vs. target (m) CpG, CHG, CHH, and CH DNA methylation levels using 10,000 reads corrected for estimated GC content ().
Nucleotide biases in genomes – such as the depletion of CpG dinucleotides to localized “CpG islands” in mammalian genomes – may interfere when estimating . CpG dinucleotides can be directly measured from 10,000 50-bp genomic sequencing reads (Table S1), and this can then be used to directly calculate the proportion of target sites that are methylated, , using the frequency of intact target sites, e.g., CpG, that remain in the bisulfite sequencing data. These are sites that were methylated and thus escaped C to T conversion. Accommodating for nucleotide biases in mammalian genomes does not improve assessment of DNA methylation levels by FASTmC (Table S1). However, treating mammals separately from other species with CpG DNA methylation (i.e., phylogenetic correction) produces an improved, mammal-specific model with similar accuracy – measured as the Mean Absolute Percentage Error (MAPE) – to the remaining species (Table S1). Additionally, only a modest increase in model improvement was observed for nonmammalian species (Table S1). Overall, GC content correction () and treating mammalian species separately improves model accuracy without introducing additional genomic sequencing data.
FASTmC also tolerates high contamination and error rates associated with sodium bisulfite conversion. We used WGBS data from A. thaliana met1 mutants (Stroud et al. 2013), which show minor (∼3%) to large (∼14%) differences in CpG DNA methylation compared to wild-type A. thaliana. By artificially introducing unmethylated chloroplast reads to 10,000 reads to met1 and met1+/− mutant genotypes, and MET1+/+ and A. thaliana wild-type genotypes, we were able to demonstrate that an ∼3% difference in DNA methylation can still be detected with < 10% chloroplast contamination, and a difference of 13%–14% with 40%–50% chloroplast contamination (Table S1). Similarly, nonconversion rates > 3% still allow for detection of differences between samples (Table S1). It should be noted that the met1 mutants and A. thaliana samples had nonconversion rates of 0.50%, 0.82%, 1.86%, and 0.56% for met1, met1+/−, MET1+/+, and wild-type A. thaliana, respectively. The artificially introduced error rates are extremely high, but possible. For example, < 1% of reads typically map to the chloroplast genome, and nonconversion rates are typically < 2% (data not shown). However, it is recommended that Lambda DNA be sequenced for each batch of WGBS libraries prepared to estimate the rate of sodium bisulfite nonconversion. Reducing technical error is especially important for identifying differences between species with small amounts of or no DNA methylation like insects (Table S1). Regardless, the FASTmC method is robust as it is able to tolerate technical and biological contamination.
The number of short-reads (≥ 30 bp) required to make accurate estimations is low, and we have determined that a few thousand reads produce high-confidence estimates of genome-wide methylation levels (Figure S1). Additionally, very little variation in predicted DNA methylation level is observed [standard error (se) = 0.0013] between 20 replicates of 10,000 randomly sampled reads (Table S1). Thus, these models can be used to accurately and cost-effectively identify differences of DNA methylation levels for any species regardless of the availability of a reference genome assembly.
Non-CpG DNA methylation can also be confidently predicted within and between species using FASTmC. In A. thaliana, the majority of DNA methylation at CHG sites is maintained by chromomethylase CMT3 through a reinforcing loop with H3K9me2 methylation catalyzed by the KRYPTONITE (KYP)/SUVH4 protein (Jackson et al. 2002; Du et al. 2012, 2014). Similarly to MET1, mutations in CMT3 cause reductions in CHG DNA methylation (Stroud et al. 2013), which are accurately detected by FASTmC (Figure 1A). Also, in A. thaliana, cell-type specific levels of CHH DNA methylation in the sperm cell (SC) (i.e., hypo-CHH DNA methylation) and vegetative nucleus (VN) (i.e., hyper-CHH DNA methylation), and depletion of CHH DNA methylation in mutants in the de novo DNA methylation pathway (e.g., the DNA-dependent RNA polymerase, POLIV) were recapitulated (Figure 1A) (Calarco et al. 2013; Stroud et al. 2013).
In mammals, non-CpG DNA methylation can be found at CH sites. A previous study demonstrated an overall increase of CH DNA methylation during brain development in M. musculus and H. sapiens (Lister et al. 2013). FASTmC was able to capture the overall trend of increasing CH methylation through brain development in H. sapiens (Figure 1A). Furthermore, despite only small differences in brain CH methylation in the intervals from 2 to 5 yr (0.068%), and from 55 to 64 yr (0.062%) of age, the FASTmC model accurately detected these changes (Figure 1A) (Lister et al. 2013).
In conclusion, we propose several models that capture the variation of, and can accurately predict, genome-wide DNA methylation levels between species to represent FASTmC, and these can be found at http://fastmc.genetics.uga.edu. Additionally, the web-based interface makes FASTmC universally accessible, and models will be continuously updated when new whole genome and methylome data are analyzed and become available. Although genome content biases interfere with the accuracy of FASTmC, treating mammalian species separately for CpG DNA methylation overcame this obstacle. FASTmC makes previously intractable studies practical (e.g., high-resolution time course, developmental, and large diversity panels) regardless of species, genome size, and availability of a reference genome. Furthermore, these models will greatly contribute to high-resolution screening of either developmentally or environmentally induced epigenomic reprogramming events. FASTmC is a suite of powerful models that can aid researchers to make better investments in more comprehensive, fruitful studies.
Acknowledgments
We would like to thank Nathan Springer for critical comments on this manuscript. We would also like to thank David Brown for webpage setup. The study was funded by grants from the National Science Foundation (MCB-1339194) and the National Institutes of Health (R00GM100000) to R.J.S. The authors declare no competing interests.
Footnotes
Supporting information is available online at www.g3journal.org/lookup/suppl/doi:10.1534/g3.115.025668/-/DC1
Communicating editor: T. R. Hughes
Literature Cited
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}