





Abstract
Standard genome-wide association studies (GWAS) scan for relationships between each of p molecular markers and a continuously distributed target trait. Typically, a marker-based matrix of genomic similarities among individuals (G) is constructed, to account more properly for the covariance structure in the linear regression model used. We show that the generalized least-squares estimator of the regression of phenotype on one or on m markers is invariant with respect to whether or not the marker(s) tested is(are) used for building G, provided variance components are unaffected by exclusion of such marker(s) from G. The result is arrived at by using a matrix expression such that one can find many inverses of genomic relationship, or of phenotypic covariance matrices, stemming from removing markers tested as fixed, but carrying out a single inversion. When eigenvectors of the genomic relationship matrix are used as regressors with fixed regression coefficients, e.g., to account for population stratification, their removal from G does matter. Removal of eigenvectors from G can have a noticeable effect on estimates of genomic and residual variances, so caution is needed. Concepts were illustrated using genomic data on 599 wheat inbred lines, with grain yield as target trait, and on close to 200 Arabidopsis thaliana accessions.
The advent of an enormous amount of DNA markers has given impetus to thousands of genome-wide association studies (GWAS) in humans, plants, and livestock (Yu et al. 2006; Manolio et al. 2009; Brachi et al. 2011; Gondro et al. 2013; Lipka et al. 2015); Neimann-Sorensen and Robertson (1961) represents one of the earliest searches for association between markers (blood groups in their study) and quantitative traits in animal genetics.
The most prevalent statistical method used in GWAS has been ordinary least-squares (OLS) linear regression of some phenotypic measurement on the number of copies of a reference allele at a single marker locus, i.e., single marker regression (SMR). This method was subsequently enhanced by use of mixed linear model methodology, originally developed in animal breeding by Henderson (1948), with the purpose of accounting for correlated observations due to genetic or genomic similarities among individuals. Ignoring such correlations, as is done in OLS or in standard logistic regression, overstates precision and creates bias in populations undergoing artificial selection (Henderson 1975). These expectations from mixed model theory were corroborated prior to the GWAS wave by Kennedy et al. (1992) using a model where individuals were genotyped for a known gene; the genetic resemblance among individuals in the sample was accommodated using a random factor (additive effects) with levels that were correlated according to the infinitesimal model. In the SMR-GWAS context, Aulchenko et al. (2007) and Meyer and Tier (2012) also used an additive infinitesimal random effect with covariance matrix proportional to a pedigree-based kinship matrix, A (Henderson 1976). Given that molecular markers have become increasingly available, it was then natural to consider replacing A by a genome-based matrix G constructed using pairwise similarities in state between individuals. Variants of this type of matrix—called genomic relationship matrices—are used widely for SNP-based analysis in animal or plant breeding and human genetics (Nejati-Javaremi et al. 1997; Yu et al. 2006; Van Raden 2008; Astle and Balding 2009; Yang et al. 2010; Price et al. 2010; Legarra 2015). Along these lines, Teyssèdre et al. (2012) used analysis and simulation to study Type I error and power behavior of four models for GWAS; their conclusion was that the performance of SMR-OLS degraded relative to generalized least-squares as heritability and variability of relationships among individuals increased.
The preceding methods were also adopted and enhanced by human geneticists. Price et al. (2010) pointed out that how best to construct G in order to perform GWAS in some optimal manner, e.g., to account for population stratification, was unclear. Consider the following question: if marker j in a GWAS is tested as a fixed effect in a SMR mixed model that includes G in the covariance structure, should the contribution of such marker to genomic similarity be removed from G? It is well understood (e.g., Goddard 2009) that if a marker effect is treated as random then it contributes to the covariance structure, but it is not a location parameter of the phenotypic distribution, since the mean vector of the latter does not depend on zero-mean random effects. Hence, if a SMR model using G treats a marker as fixed and tests for its effect, the test would not appear to be net, as the marker is also contributing to variance, i.e., it is implicitly included in the model as a random effect. In other words, the marker is viewed as having a fixed and a random effect simultaneously. The contradiction is clear: if a marker effect is a fixed parameter, it cannot have a frequentist variance. Consider a GWAS scan involving p markers; if the marker to be tested were to be removed when building up G, p distinct genomic relationship or phenotypic covariance matrices (of size n × n each) would need to be constructed and inverted (depending on the algorithm). The analysis would be impractical if the number of variants tested is large, as is the case with sequence data. It is fairly obvious that if p is very large, the impact of removing a marker should be nil. However, the question posed above deserves an unambiguous answer.
A similar issue arises in many treatments of genome-derived population structure presented in the literature. In principle, substructure must be accounted for in a GWAS somehow so that the analysis informs about association in a conceptually homogeneous population (Yu et al. 2006; Zhu and Yu 2009). Yang et al. (2010, 2011) accounted for population structure by extracting principal components (PC) from G, and regressions on a subset of these were regarded as fixed in a mixed model, but with G used without any modification; Stahl et al. (2012) presents an application. Janss et al. (2012) argued that such an approach would be ill-posed because it produces double counting: the eigenvectors of G used as covariates in the fixed part of the model are also an implicit part of G. Should G then be left intact? On one hand, the view could be taken that if an eigenvector is used as a covariate with a fixed regression coefficient, its contribution to G should be discounted. On the other hand, removal of the eigenvector could degrade the measure of similarity among individuals. Janss et al. (2012) pointed out that the eigen-decomposition of G would provide a solution to the problem (attenuation via eigenvalues) when the aim is to infer marker effects and genomic heritability. However, such attenuation shrinks all regressions on eigenvectors to 0 (to distinct degrees), and shrinkage on regressions on markers with medium or large effect sizes, or on eigenvectors used to account for population stratification, should not be exerted. Under such reasoning, the contribution to G of a marker or of eigenvector(s) associated with fixed regressions, should be discounted if one seeks net effect size estimates and corresponding tests of hypotheses. Another view (Astle and Balding 2009; Rincent et al. 2014) is that G conveys information on both population structure and relatedness, so it may “not be useful to consider admixture information as fixed effects covariates.” The preceding discussion reflects a lack of consensus in the GWAS field.
In this paper, we address the construction of G in a GWAS context. First, we describe how a single marker GWAS with, say, p conveniently constructed G matrices, can be carried out using a mixed linear model in which the marker effect tested is treated as fixed and the remaining p − 1 markers are used to introduce similarities in state, i.e., p − 1 markers are viewed as having zero-mean random effects. A similar approach is discussed for the situation where eigenvectors are chosen as regressors (with fixed regression coefficients), with the purpose of accounting for population stratification, with the remaining eigenvectors used to induce similarities among individuals. In particular, we show algebraically that the generalized least-squares estimator of the regression on a marker is invariant with respect to whether or not the marker (or set of markers) treated as fixed is used when building G. It is also shown that removal of eigenvectors does matter. The manuscript is organized as follows. Basic concepts of GWAS conducted with kinship matrices in the model are reviewed in the sections under Standard Approaches Used in GWAS. Next, in Impact of Removing Markers from the G-Matrix, it is shown how inverses of the p needed genomic relationship (or of phenotypic covariance) matrices can be found in a convenient manner, and use the results to prove the invariance indicated above; removal of eigenvectors is also discussed in this section. To illustrate main, as well as related, concepts, data on wheat inbred lines and on A. thaliana were used, as described in Case Studies with Wheat and Arabidopsis Data. Technical details are shown in Appendices to the paper and toy examples are in Supplemental Material, File S1.
Standard Approaches Used in GWAS
Ordinary least-squares SMR
Generalized least-squares (GLS) SMR using a matrix of realized genomic relationships
Impact of Removing Markers from the G-Matrix
General considerations
As pointed out earlier, if a regression on a marker is treated as a fixed effect, it would seem sensible to remove its contribution to . Otherwise, there would be a contradiction: a fixed effect affects the mean of a distribution but does not contribute to covariance structure. Conversely, a zero-mean random effect contributes to dispersion (variance and covariance) but not to location (e.g., Henderson 1984; Gianola 2013)
Conceivably, a marker effect could be modeled as the sum of a fixed and of a random component; the fixed part would index the mean of the distribution, and the random part would contribute to the likelihood only through covariance structure. This view, however, contradicts quantitative genetic theory, where quantitative trait locus (QTL) effects are fixed and genotypes are random (e.g., Falconer and Mackay 1996; Lynch and Walsh 1998; Gianola et al. 2009; Gianola 2013; de los Campos et al. 2015). In classical quantitative genetics, QTL effects do not have variance but these loci generate variance in allelic content among individuals. On the other hand, Bayesian regression models pose a variance on effects that reflects uncertainty, a priori. Actually, the Bayesian treatment of a fixed effect (e.g., a flat prior) implies an infinite prior variance provided the flat prior is unbounded. Here, we examine the question of whether or not a marker effect declared as fixed can also be allowed to have a random effect, as implied when including the marker in question in the building of G.
GWAS using the GLS representation
If the effect of marker j is fixed in the GWAS, and the marker is removed when constructing G, p distinct genomic relationship matrices need to be built to carry out the GLS GWAS, accordingly, with p or matrices formed and inverted. This procedure is computationally taxing if p is large. A short-cut is described below, with the result used subsequently to show that the GWAS can actually be carried out using without modification.
GWAS and BLUP using the mixed model representation
It is concluded that point estimates and point predictions from GLS and BLUP respectively, are invariant with respect to whether or not the marker being tested as a fixed effect is included or removed when constructing the type of genomic relationship matrix used here.
Generalizations
Several markers tested as fixed effects simultaneously:
Removing eigenvectors from G:
Case Studies with Wheat and Arabidopsis Data
Wheat
A publicly available wheat data set was employed to investigate several issues associated with removing markers or eigenvectors from including impact on maximum likelihood estimates of variance components. The wheat data were downloaded from package BGLR (Pérez and de los Campos 2014); these data have also been used by, e.g., Crossa et al. (2010), Gianola et al. (2011) and Long et al. (2011). The data originated from several international trials conducted at the International Maize and Wheat Improvement Center (CIMMYT), Mexico. There are 599 wheat inbred lines, each genotyped with 1279 DArT (Diversity Array Technology) markers and planted in four environments. The target trait was yield in environment 1. Here and . The DArT markers are binary denoting presence or absence of an allele at a marker locus in a given line. In this data set there is no information on chromosomal location of markers, but this does not hamper illustration of concepts.
Arabidopsis
We also used the A. thaliana data set described by Atwell et al. (2010) and Wimmer et al. (2013), mainly for illustrating the impacts of eigenvector removal on inferences. Norborg et al. (2005) and Atwell et al. (2010) pointed out that this sample of accessions suggests a complex structure in the population, making the data interesting for our purposes. The data, available in the R Synbreed package (Wimmer et al. 2012), represents 199 accessions genotyped with a custom Affymetrix 250K SNP chip, and measured for a number of phenotypes. As in Wimmer et al. (2013), flowering time , plant diameter , and FRIGIDA gene expression were chosen as target phenotyes; marker genotypes are pre-edited in the package, and 215,947 SNP loci were used in the analysis.
GWAS: OLS vs. GLS analyses
We compared SMR-OLS vs. GLS in the wheat data at two specified values of the variance ratio or, equivalently, of genomic heritability. Marker genotypes were centered to have a mean of zero, marker by marker, for all 1279 DArT polymorphisms; phenotypes were already standardized to have a null mean and variance 1. For OLS, computation was done using the lm function available in the R package (http://www.r-project.org/). In GLS, the genomic relationship matrix used was as follows: 1) with being the matrix of centered markers, we formed ; here, is the mean of the diagonal values of and measures similarity in state between individuals i and j. 2) We then formed , and, for the purpose of examining sensitivity, set genomic heritability in the GLS analysis to representing an increase in the signal to noise ratio when going from 0.10 to 0.25. GLS was implemented using the lm function via transformation of the phenotypes and of the marker incidence matrix, as shown in Appendix D.
The SMR OLS and GLS analyses gave similar inferences in terms of regression coefficients, (percentage of corrected sums of squares of grain yield explained by the model), and values, but the GLS residual variances were smaller. While 29 markers were found significant (Bonferroni corrected p values) for OLS, the GLS analyses at and 0.25 produced 32 significances (31 in common). As is typically the case for quantitative traits such as grain yield, most single marker based models explained a small fraction of the variation: the largest observed were 7.27%, 7.28%, and 7.29% for OLS, GLS(0.10), and GLS(0.25), respectively. Here, was the standard measure used in OLS and GLS (using Appendix D, one can calculate the GLS statistics employing OLS computations); alternative measures are discussed by Sun et al. (2010).
The GWAS literature does not emphasize enough that the explanatory power of a model and estimates of effect sizes change markedly when additional markers are included in the specification, i.e., failure to account for other variants is one of the most obvious explanations of missing heritability (Maher 2008) in SMR. Adding up from SMR gives a distorted picture of the variability explained, because LD is ignored (e.g., Gianola et al. 2013). To illustrate how effect size in GWAS was affected by model specification, we fitted jointly by least-squares multiple marker regression (MMR) all 29 markers found significant in OLS SMR. The of this model was 28.3%. In this MMR, however, only two markers were significant at (Bonferroni correction); these p values are of course incorrect in a sequential approach such as the one followed here. Effect size estimates were different, including sign changes (some markers with a negative SMR estimate became positive in MMR, and vice versa) The MMR estimates were larger in size and more variable (SE not shown) due to the colinearity caused by strong LD between some markers. In theory, SMR may have a larger bias (relative to causal loci in a multifactorial model) than MMR, but the latter produces estimates with more variability. The mean-squared error of estimation cannot be evaluated in the absence of knowledge of model parameters; the true marker effect depends on the effects of the QTL affecting the trait, and on the unknown LD relationships between markers and QTL, these being of a multivariate nature in the case of complex traits (de los Campos et al. 2015)
Effect of removing a single marker from G on genomic heritability
Since our analytical developments assume that marker removal does not affect the partition of variance, we measured the extent to which this assumption held using the wheat data set. The likelihood was formed under . Here, we took , where is the mean of the diagonal elements of (markers were centered) and estimated the two variance components by maximum likelihood using an eigen-decomposition algorithm of (e.g., Janss et al. 2012) that renders computation fast. Estimates obtained with all markers included in were and ; genomic heritability was 0.529. Convergence was assessed and confirmed by beginning the iteration from different sets of starting values. Further, we constructed 1279 genomic relationship matrices by excluding one marker at a time, i.e., , convergence was assumed for each case, starting the iteration using a value of 0.5 for each of the two variance components. The estimates obtained are shown in Figure 1; larger estimates of genomic variance were associated with smaller estimates of residual variance. Departure of parameter estimates from the values obtained when all markers entered into G was very mild for all markers. Out of the 1279 sets of estimates of genomic and residual variance, 797 were smaller than due to marker exclusion.
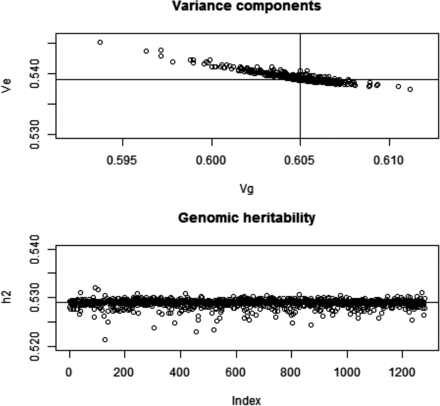
Wheat: maximum likelihood (ML) estimates of genomic (Vg) and residual (Ve) variance components and of genomic heritability (h2) corresponding to 1279 models with markers removed, one at a time, when forming the genomic relationship matrix (G). Top panel: variance components; horizontal and vertical lines indicate ML estimates with all markers in G. Bottom panel: genomic heritability; horizontal line indicates the estimate with all markers.
We assessed genetic variability as often done in GWAS via SMR, and related the corresponding metrics to what would be suggested by a variance component analysis. In a SMR, the contribution of marker j to variability is calculated as where is typically an OLS estimate and q is allelic frequency; stands for single marker variance. This formula is very crude because it assumes that the trait is mono-factorial, or that loci are in linkage equilibrium, so the total genetic variance would be the sum of variances contributed by each of the loci (e.g., Gianola et al. 2009, 2013). For inbred lines such as in the wheat data, the metric is We evaluated if changes in estimates of due to removing marker j from the G matrix correlated with , and with standard values from SMR. Since marker removal sometimes increased, sometimes decreased, the variance estimates relative to we computed the absolute values of ; Figure 2 displays relationships between , and . SMV and had a clear association; this was expected because is proportional to in simple linear regression. Since the relationship between Δ-V and SMV, or , was less transparent, we extracted a pattern using local regression (LOESS) with a span parameter of 0.25, meaning that a local neighborhood had 320 members (Cleveland 1979). There was a tendency for Δ-V to increase when SMV (or increased. This is reasonable because the more variance a marker captures, the larger the decrease from should be when such marker is removed from G. Since we were unable to monitor convergence for the 1279 sets of estimates, it may be that removing a marker increased relative to ; this phenomenon could be due to convergence to a local maximum and our estimation procedure did not constrain each to be, at most, Thus, we turned attention to the subset of estimates where marker removal reduced the marked additive genetic variance relative to . This analysis is displayed in Figure 3 and the picture was clear: removing markers from G assessed via SMR as making a larger contribution to the variance of the trait did reduce estimates of genomic variance. The impact was very small but detectable.
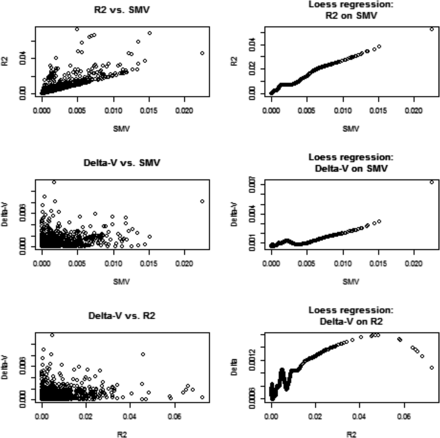
Wheat: associations between change in absolute value of genomic variance estimate due to removal of a marker (Δ‐V), and the R2 and marker variance assessment (SMV) from single marker regression. Right panels show fitted values of a local regression (LOESS) with span parameter = 0.25.
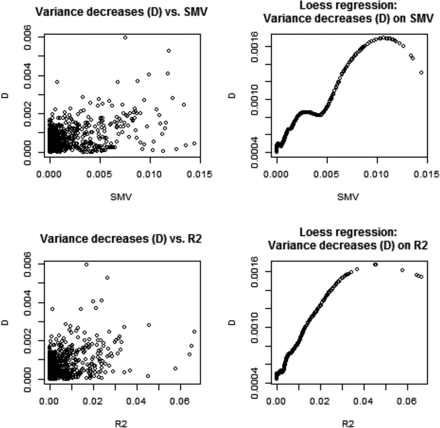
Wheat: associations between decrease (D) in genomic variance estimate due to removal of a marker from the genomic relationship matrix (G), and the R2 and marker variance assessment (SMV) from single marker regression. Plot depicts the 482 cases where marker removal reduced the genomic variance estimate relative to the estimate obtained with all markers contributing to G. LOESS span parameter = 0.40.
It is concluded from the preceding that, if the contribution of a marker to G is removed, it is safe (especially with dense chips) to use variance components estimated from an all-markers analysis, unless there are some huge effect variants that would probably be detected anyhow. Since we showed analytically that the GLS estimator is invariant with respect to removing markers from G, it is unnecessary to re-estimate variance parameters, or to modify G at any pass of a SMR GWAS.
Effect of removing principal components from G on genomic heritability
Multi-dimensional scaling of wheat and Arabidopsis:
Many GWAS use a SMR model with one or a few regressions on PC of as fixed effects, to account for population structure. If such an analysis is based on a mixed model with as genomic covariance structure, the implication is that the PC fitted (with a fixed regression) is not removed from G, which is contradictory. To guide the specification of the GWAS model, we searched for genomic structure in the wheat and Arabidopsis genotype matrices using multi-dimensional scaling (MDS).
MDS was developed by Kruskal (1964 a, b) to obtain spatial representations of objects in a perceptual dimensional topology. A description is in Borg and Groenen (2005), and an application to quantitative genomics is in Zhu and Yu (2009). MDS inputs can be squared Euclidean distances between objects, in our case the dimensional genotypes of the 599 wheat lines or the 199 Arabidopsis accessions. There were 179,101 and 19,701 Euclidean distances between rows of X (or of G) in the wheat and Arabidopsis data sets, respectively. In MDS, distances are rotated into a matrix whose eigen-decomposition yields the K-dimensional coordinates, while preserving distances in some best fit sense. There are two types of MDS: classical and nonmetric. In classical MDS, differences and ratios between distances are preserved. In nonmetric scaling, only the order of the distances is relevant. The best fitting K is found at the eigenvalue in which an elbow of an eigenvalue decay plot is observed, or can be derived from a metric called STRESS. Here, squared differences between observed and fitted distances are summed over the two dimensions, and expressed relative to the sum of all observed squared distances. If STRESS (the square root of the preceding quantity) is smaller than 5–10%, the corresponding dimension is deemed to give a satisfactory fit (Kruskal, 1964b).
We fed the wheat and Arabidopsis distances to the R functions cmdscale and ISOmds, fitted models with (Arabidopsis) and (wheat) dimensions, and calculated STRESS for each model. Spatial representations obtained for models with two dimensions are in Figure 4: the perception of structure is much clearer in wheat than in Arabidopsis. In wheat, the first coordinate separates lines into two well delineated groups; the second coordinate stretches the lines within groups along the y-axis. A two-dimensional representation seemed insufficient in the Arabidopsis data. Figure 5 (top panel) presents a scree plot of eigenvalues expressed as a fraction of the sum of all MDS eigenvalues: the first five eigenvalues represented 31.8% and 14.5% of the variation in wheat and Arabidopsis, respectively; save for the first two, there were no clearly dominant values in Arabidopsis. The middle and bottom panels show STRESS (nonmetric MDS) for models of different dimensionality. In the wheat data set, a satisfactory fit (STRESS = 5–10%; Kruskal 1964b) was obtained with dimensions, but at least 70 dimensions were needed to fit the Arabidopsis distances reasonably well. The implication is that population structure in wheat may not be accounted properly with a single PC. In the Arabidopsis collection, the topology was less sharp, suggesting an aggregation similar to the family structure typically encountered in animal breeding or in humans (see Supplementary Figure 4 in Atwell et al. 2010). If the latter is the case, use of a kinship matrix in the GWAS may suffice, without the need of fitting principal components as fixed effects. In wheat, on the other hand, a kinship matrix would account for similarity among lines, but not for differences in mean among the few strata suggested by Figure 4 and Figure 5.
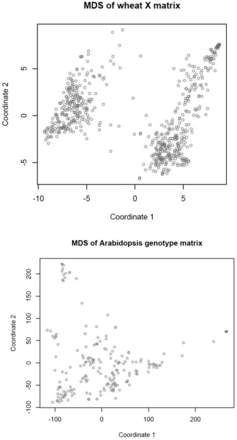
Multidimensional scaling of SNP genotype matrices in the wheat and Arabidopsis data sets: first two dimensions.
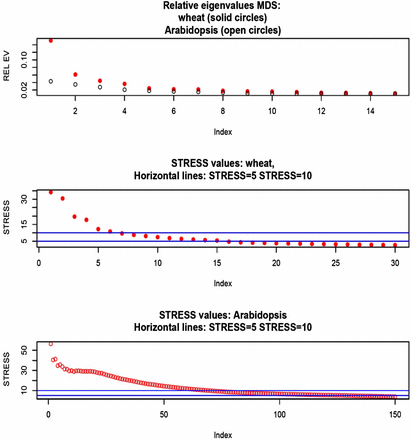
Multidimensional scaling of SNP genotype matrices in the wheat and Arabidopsis data sets. Top panel: eigenvalue (relative to their sum) decay. Middle panel: STRESS metric in wheat. Bottom panel: STRESS metric in Arabidopsis.
PC and variance components:
We examined the effect of removing PC from on maximum likelihood estimates of the two variance components. In wheat, after extracting the 599 PC of G, maximum likelihood estimates of and were obtained by removing one PC at each time when building the genomic relationship matrix. The impact on the estimates was noticeable (Figure 6): removing “dominant” PC from produced much lower estimates of genomic variance and of genomic heritability than when all PCs entered into G (recall that genomic variance and heritability of yield were 0.605 and 0.529, respectively), and larger estimates of residual variance. The relationship between (genomic antiheritability) and the from the OLS regression of grain yield on each of the PC is shown in Figure 7: removal of PC with the largest resulted in the largest antiheritability estimates. PC with the strongest association with the trait also had the largest impact on estimates when removed from G (not shown) In short, if a PC is treated as fixed but not removed from G, the residual variance will be understated, because the fact that such PC also contributes to genomic variance would not be accounted for properly. Genomic heritability should be re-estimated if PCs are removed from G.
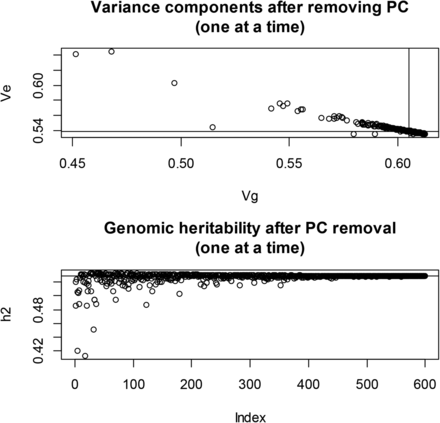
Wheat: maximum likelihood estimates of genomic (Vg) and residual (Ve) variance components and of genomic heritability (h2) corresponding to 599 models with principal components (PC) removed, one at a time, when forming the genomic relationship matrix (G). Top panel: variance components. Bottom panel: genomic heritability. Horizontal and vertical lines indicate estimates found with all PC in G.
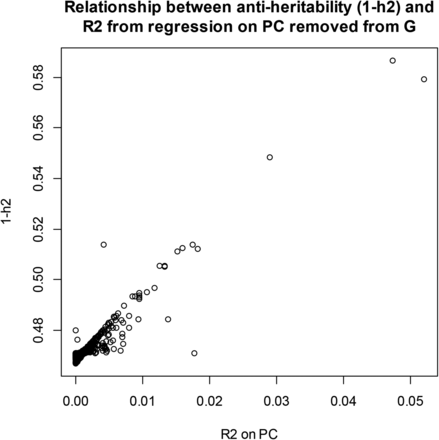
Wheat: relationship between genomic antiheritability (1–h2) after removing each one of the PC of G and R2 from the ordinary least‐squares (OLS) regression of yield on each of the PC.
In Arabidopsis, estimates of variance components and of genomic heritability were , and (flowering time); , and (FRIGIDA), and , and (plant diameter). Close to 50% (FRIGIDA and diameter) and near 100% (flowering time) of the variance among accessions was accounted for by the 215,947 markers used for building the genomic relationship matrices. However, SE of the estimates were very large, reflecting the small number of accessions in the sample. Nevertheless, the heritability estimate of flowering time suggests a large degree of genetic control of the trait. Figure 8 displays effects on the dispersion parameters of removing PC from the genomic relationship matrix. In general, removing any of the first 10–30 PC had the largest impact on the decrease of genomic variance and heritability, and the concomitant increase in residual variance, particularly for flowering time. There were exceptions, however; for instance, removing PC 4 had a similar effect on the decrease of genomic variance than removing PC 100 or PC 110. A larger sample size would probably produce a more discernible pattern.
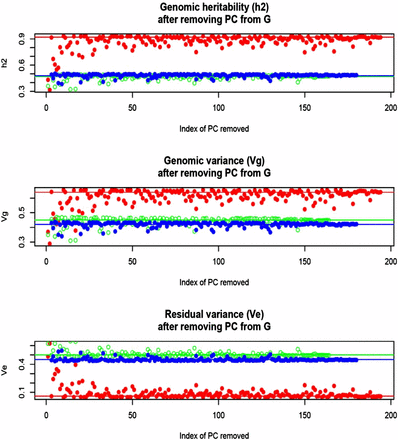
Arabidopsis: maximum likelihood estimates of genomic (Vg) and residual (Ve) variance components and of genomic heritability (h2) corresponding to models with PC removed, one at a time, when forming the genomic relationship matrix (G). Red: flowering time. Green: FRIGIDA expression. Blue: plant diameter. Horizontal and lines indicate estimates found with all PC in G.
PC in the regression model:
We evaluated the extent to which associations detected by OLS or GLS were affected by accounting for structure, and by whether or not the PC used as regressor was kept as a part of, or excluded from, the genomic relationship matrix G. With that objective, we compared estimates from various analyses of the wheat data: 1) OLS on markers with and without the first PC as a covariate; 2) GLS (using maximum likelihood estimates of variance components) on markers with or without the first PC as covariate; 3) GLS as in (2) but with or without the first PC removed from the G matrix. As expected, increased, while some regressions near 0 became more negative and some more positive when the PC was included in the model; this happened both in OLS and GLS. Several regressions on markers became more significant because the residual variance decreased relative to the one produced by the model without the PC as regressor. Including or excluding the first PC when forming the relationship matrix G had a negligible impact on inference, as the metrics used in the comparison aligned on a 45° degree line (results not shown).
In Arabidopsis, we fitted a multiple-regression on the first 70 PC; this was done by OLS and by GLS (maximum likelihood estimates of variance components, all PC in G). The support for statistical significance is shown in Figure 9. For flowering time, nine (eight) of the 70 regressions were declared significant by GLS (OLS). For FRIGIDA, only one regression was deemed significant by GLS, and none for OLS. For plant diameter, the two methods agreed. The analyses illustrate that the effects of population structure are trait-dependent. For flowering time, the trait with the largest relative amount of genetic variance, several PC seem needed for an appropriate GWAS; ignoring this complex structure could provide a false idea of association expected within a homogeneous group of accessions.
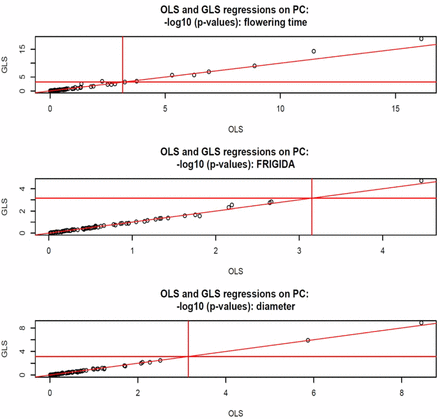
Arabidopsis: OLS and generalized least‐squares statistical support for association with 70 principal components of G fitted jointly, –log (p‐values, base 10), for flowering time, FRIGIDA expression, and plant diameter. Horizontal and vertical lines are at 3.15, corresponding to a Bonferroni correction for 70 comparisons with single test significance at 5%.
We extracted the first 5000 markers from Arabidopsis chromosome 2, and used flowering time (the trait with 90% heritability) as a target trait for evaluating alternative GWAS model specifications. Genomic relationship matrices were constructed with all 215,947 markers, and the regression model included the single marker tested, and either zero, one, five, 10, or 50 PC as fixed covariates. Figure 10 gives a plot of allelic substitution effects, and of : the x-axis labels effect size estimates (top panel) and statistical support values (bottom panel) for the model without PC in the regression structure. Clearly, accounting for structure had a marked effect on estimates of marker effects, and on statistical support: as the number of PC in the regression increased, effect sizes decreased in absolute value and support for association vanished. While a large number of markers would be declared as associated when population structure is ignored, only a few of these would remain significant after the first PC is fitted; none would be significant if five or more PC are fitted. When the first PC was removed from G, heritability of flowering time dropped from 0.92 to 0.43. When five, 10 or 50 PC were removed, decreased further to 0.07, and It is interesting to note that, while the nonmetric MDS suggested that about 50 dimensions were needed to account for genomic dissimilarity among accessions, only a few dimensions capture the association with trait variance.
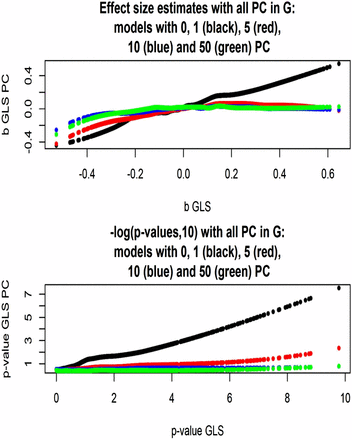
Arabidopsis: effect sizes and statistical support for association between flowering time and 5000 markers (chromosome 2) for models without or with one, five, 10, or 50 PC as fixed covariates. Models use a genomic relationship matrix with all its PC and corresponding maximum likelihood estimates of variance components. Scatter was smoothed using LOESS.
Finally, effect size estimates and statistical support were compared for the model in which G was left intact when one, five, 10, or 50 PC were fitted in the regression structure, vs. the corresponding models with G and variance components appropriately modified. As shown in Figure 11 (top panel), effect size estimates aligned well for the two classes of model, but their absolute values were somewhat smaller when the contribution to G of the PC tested was taken into consideration. The bottom panel of Figure 11 shows that the statistical support for association essentially vanished when more than two dimensions of the population structure were accounted for via PC.
![Arabidopsis: effect sizes and statistical support [–log(p‐value, 10)] for association between flowering time and 5000 markers (chromosome 2) for models without or with one, five, 10, or 50 PC tested as fixed covariates. Models use a genomic relationship matrix with or without the PC tested included, and corresponding maximum likelihood estimates of variance components.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/g3journal/6/10/10.1534_g3.116.034256/8/m_3241f11.gif?Expires=1716563402&Signature=SPMubd8KILFKrXTtOqoeMDet2kHc2Tlh1p7Un78M1EyontHSo~chdqMJewl5PzX223GwCEA-wJq9DO~cseH1VjQD8Fu1MGboWT2GUFHW4Ek3jZdf5Efu1GA6VIxZrv29CdmlVOiyDpvvRmyeRzFwY-jum76d-0azwvYQ0cxtGbAx5c0ZvBfsLSje4S0wQ8gWFj3GWA~D2YH5UlyNFgFWGDI3lHDI-H1h~CZpJ6Qdpqz-Z2LV6hz8ux2DI2aQrNVo3WBQGto4bD2y14XyIL7oFE~yXfDAyEXgMOjxzYBvf0bmHpMzJgr-jRLzXOmUU6yk0bmhInDKYNjLEy9vohV60w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Arabidopsis: effect sizes and statistical support [–log(p‐value, 10)] for association between flowering time and 5000 markers (chromosome 2) for models without or with one, five, 10, or 50 PC tested as fixed covariates. Models use a genomic relationship matrix with or without the PC tested included, and corresponding maximum likelihood estimates of variance components.
Data availability
The authors state that all data necessary for confirming the conclusions presented in the article are represented fully within the article.
Conclusions
Our study addressed some standing issues in standard GWAS methodology for complex traits, as practiced in animal, human, and plant genetics. We examined the question of how removal of one or more markers from the genomic relationship matrix affects the generalized least-squares estimator (maximum likelihood under normality, and known genomic heritability) of allelic substitution effects in a SMR. It was shown analytically that, if variance components are kept constant, the GLS estimator and the GBLUP predictor of marker additive genetic values are invariant with respect to whether or not the marker(s) tested for association is(are) included when constructing G. We also examined the impact of removing PC from G, and found that it does matter, and importantly so. Further, and unsurprisingly, estimates of genomic and residual variances were found to be sensitive with respect to the structure of G. Concepts were illustrated using publicly available wheat and Arabidopsis data sets.
In conclusion, in a homogeneous population, inferences from a GWAS using GLS where the genomic relationship matrix is constructed using all markers does not present clear pitfalls other than the inability of a SMR model to represent the statistical genetic architecture of a complex trait properly. On the other hand, if one or more PC are used as fixed regressors to account for population stratification, the genomic relationship matrix perhaps should be modified, and variance components re-estimated accordingly. In the absence of knowledge of the state of nature, it is impossible to answer unambiguously the question of which approach is best. It has been argued and shown that statistical significance and predictive ability are not synonymous (Lo et al. 2015), so perhaps cross-validation could be used for comparing models. An unfortunate duality is that predictive performance does not necessarily provide a guide for explanation (Shmueli 2010).
Appendix A: Sherman-Morrison-Woodbury Formula
Appendix B: Difference Between GLS Estimators
Appendix C: Difference Between BLUP Predictors
Appendix D: Computation of GLS with OLS
Acknowledgments
Gustavo de los Campos is thanked for providing the function for maximum likelihood estimation of variance components using an eigen-decomposition. Part of this work was done while D.G. was a Hans Fischer Fellow at the Institute of Advanced Study, Technical University of Munich, Germany and a Visiting Scientist at the Institut Pasteur de Montevideo, Uruguay. Research was partially supported by a United States Department of Agriculture Hatch grant (142-PRJ63CV) to D.G., by project URUGENOMES, ATN/KK-14584-UR funded by Inter-American Development Bank, and by the Wisconsin Agriculture Experiment Station. M.I.F. was supported by Agencia Nacional de Investigación e Innovación, Uruguay, project PD_NAC_2013_10964. This work was supported by the German Research Foundation and the Technical University of Munich in the framework of the Open Access Publishing Program.
Footnotes
Communicating editor: D. J. de Koning
Supplemental material is available online at www.g3journal.org/lookup/suppl/doi:10.1534/g3.116.034256/-/DC1
Literature Cited
Henderson, C. R., 1948 Estimation of general, specific and maternal combining ability in crosses among inbred lines of swine. Ph.D. Thesis, Iowa State University, Iowa.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}