




Abstract
Population adaptation to strong selection can occur through the sequential or parallel accumulation of competing beneficial mutations. The dynamics, diversity, and rate of fixation of beneficial mutations within and between populations are still poorly understood. To study how the mutational landscape varies across populations during adaptation, we performed experimental evolution on seven parallel populations of Saccharomyces cerevisiae continuously cultured in limiting sulfate medium. By combining quantitative polymerase chain reaction, array comparative genomic hybridization, restriction digestion and contour-clamped homogeneous electric field gel electrophoresis, and whole-genome sequencing, we followed the trajectory of evolution to determine the identity and fate of beneficial mutations. During a period of 200 generations, the yeast populations displayed parallel evolutionary dynamics that were driven by the coexistence of independent beneficial mutations. Selective amplifications rapidly evolved under this selection pressure, in particular common inverted amplifications containing the sulfate transporter gene SUL1. Compared with single clones, detailed analysis of the populations uncovers a greater complexity whereby multiple subpopulations arise and compete despite a strong selection. The most common evolutionary adaptation to strong selection in these populations grown in sulfate limitation is determined by clonal interference, with adaptive variants both persisting and replacing one another.
Adaptive evolution in asexual populations depends on the accumulation of genetic variation (Gerrish and Lenski 1998). If a single beneficial mutation occurs in a small population and is not lost from drift, its frequency will increase over time to eventually reach fixation (review in Burke 2012). In larger populations, multiple beneficial mutations can occur and interfere with one another’s fixation, a phenomenon referred to as ”clonal interference” (Burke 2012). In a population in which two beneficial mutations compete, the one conferring greater fitness is more likely to reach fixation (Gerrish and Lenski 1998).
The dynamics by which mutations accumulate within populations are complex as the result of stochastic mutational processes, drift, natural selection, and competition between clones of different overall fitness (Gerrish and Lenski 1998; de Visser et al. 1999; Desai et al. 2007; Miller et al. 2011; Lang et al. 2013; Lee and Marx 2013). Such dynamics can be examined directly by laboratory experimental evolution in microbial populations. Previous long-term studies have contributed to our understanding of genetic and genomic changes that underlie diverse phenotypes (Andersson et al. 1998; Koszul et al. 2004; Cakar et al. 2005; van Maris et al. 2007; Guimaraes et al. 2008; Kugelberg et al. 2010; Adamo et al. 2012). These studies have primarily focused on clones isolated either at particular times (Sonti and Roth 1989; Notley-McRobb and Ferenci 2000; Barrick et al. 2009) or at the end of the experiments (Brown et al. 1998; Dunham et al. 2002; Gresham et al. 2008; Lee and Marx 2012), and therefore provide limited information about population dynamics. One effective way to understand the dynamics of a population undergoing adaptation is to mark different subpopulations and visualize their change in frequency over time. Neutral fluorescent markers, for example, have been used to monitor the contractions and expansions of subpopulations over 500 generations of evolution (Kao and Sherlock 2008). However, even within these defined subpopulations, multiple beneficial mutations can arise over the course of the evolution experiment, making it difficult to track the extent of clonal interference. A recent study has quantified the temporal dynamics of point mutations over 1000 generations by deep sequencing of populations (Lang et al. 2013). Similar work has been conducted by Lee and Marx in which they examined large deletions and other chromosome rearrangements during the long-term experimental evolution of Methylobacterium extorquens (Lee and Marx 2012, 2013). These studies were able to detect up to 17 different large-scale rearrangements in one population.
In a chemostat, large populations of cells grow under a controlled environment for many generations. This system allows us to study adaptation under a defined selective pressure, such as limitation for a nutrient. In both bacteria and yeast grown under nutrient limitations, the target of selection is often a nutrient transport pathway. For example, mutations in ompF, a gene implicated in membrane permeability, have been isolated in Escherichia coli grown in lactose-limited conditions (Zhang and Ferenci 1999). In Saccharomyces cerevisiae grown in glucose-, nitrogen-, or sulfate-limited chemostats, amplifications of the glucose, amino acid, and sulfate transporters (HXT6/HXT7, GAP1, and SUL1, respectively) were detected in single clones (Brown et al. 1998; Gresham et al. 2008; Kao and Sherlock 2008; Gresham et al. 2010). Because amplification of SUL1 occurs repeatedly in independent evolution experiments and confers a large fitness advantage (Gresham et al. 2008), sulfate-limited chemostat growth provides an excellent model for visualizing the diversity and dynamics of beneficial mutations. Independent mutations affecting the same gene, often called parallelism, have been previously described at the single nucleotide level (Tenaillon et al. 2012; Herron and Doebeli 2013; Lang et al. 2013) as well as for large and small deletions, amplifications, and insertions (Zhong et al. 2004; Stoebel et al. 2009; Blount et al. 2012; Chou and Marx 2012; Lee and Marx 2012). In this work, we determined that the selection for amplification of SUL1 is highly repeatable and provides an excellent example of parallelism at the locus level. We had previously isolated several independently evolved clones in which each amplification event appeared to produce novel junctions on chromosome II (Gresham et al. 2008; Araya et al. 2010), leading us to hypothesize that these unique junctions could be used to identify distinct subpopulations. We now demonstrate that the inverted repeat structure we identified previously in a single clone (Araya et al. 2010) is the dominant mode of amplification of SUL1 in haploid yeast. Unlike with the amplifications of HXT6/HXT7 and GAP1, which recur using the same repeat elements and are thus difficult to track, each SUL1 amplicon resulted in novel junctions, allowing us to use them as intrinsic markers to follow the trajectory and determine the fate of unique amplifications during the course of ~200 generations in multiple parallel independent cultures. Whole-genome sequencing of several evolved clones also identified a beneficial point mutation with a fitness effect less than that conferred by the amplification of SUL1. Tracking of multiple subpopulations provides evidence that parallel evolution via clonal interference is the mode of action by which evolutionary adaptation occurs in populations of S. cerevisiae subjected to strong selection for assimilating limiting sulfate.
Materials and Methods
Strains and continuous culture
The S. cerevisiae wild-type strain used in this study was FY4, a MATa prototroph of the S288c background. A single colony was inoculated into sulfate-limited chemostat medium (Gresham et al. 2008), grown overnight at 30°, and 100 μL of the culture was inoculated into ministat chambers (Miller et al. 2013) containing 20 mL of the same medium. After 30 hr, the flow of medium was turned on at a dilution rate of 0.17 ± 0.01 hr−1. Seven chemostats were inoculated in total and cell samples (glycerol stock and dry pellet) were passively collected every day from fresh effluent for ~200 generations. The strain used in the competition experiments is a FY MATa strain where the HO locus had been replaced with eGFP. To test the fitness due to the amplification of SUL1, we transformed ura3 strains with a low-copy plasmid (Ho et al. 2009) or a 2-μm plasmid containing SUL1 (Cherest et al. 1997). Clones at generation approximately 50, 100, and 200 were plated from frozen samples onto sulfate-limiting plates and propagated in sulfate-limiting liquid medium to attempt to maintain selection for the amplicons that arose during chemostat growth.
The strain deleted for SGF73 was obtained from the Yeast Deletion Collection (MATa sgf73::KanMX his3Δ1 leu2Δ0 lys2D0 ura3Δ0) (Giaever et al. 2002). The strain was backcrossed three times to FY5 (MATα, prototroph) to select for a prototroph clone that contained the sgf73 deletion.
Genomic DNA extraction, gel electrophoresis assays
Genomic DNA was extracted from dry, frozen cell pellets via the Smash-and-Grab method (Hoffman and Winston 1987) or the NIB-and-Grab method, which is a hybrid of the Smash-and-Grab procedure and Huberman DNA isolation procedure (Huberman et al. 1987; see Supporting Information, File S1). The average molecular weights of the two DNA isolation methods yielded DNAs of 10−20 kb and >50 kb, respectively. Gels for analysis of restriction enzyme digested genomic NIB-and-Grab DNA were 20 cm 0.4% ME agarose run at 1−1.5 V/cm for 18−24 hr in 1X TBE. The probes for Southern blot hybridization were an internal fragment of SUL1, a fragment just centromere proximal to SUL1 (“786”), a fragment of chromosome III containing ARS305, a fragment from chromosome V containing ARS522 (originally known as ARS501), and the CEN2 adjacent ORF ECM15.
For indirect end labeling NIB-and-Grab DNAs were digested first with either ApaLI, which cleaves just centromere-proximal of the 5′-end of the SUL1 gene with no additional ApaLI sites between SUL1 and the right telomere (~25 kb), or EcoNI, which releases a fragment that extends from the 3′ end of SUL1 19.4 kb toward CEN2. Aliquots (~1 μL) of these digests were incubated with a series of second enzymes that cleave varying distances from SUL1. For snap-back assays, NIB-and-Grab DNA was digested with either ApaLI or EcoNI, denatured at 100° for 10 min, chilled immediately on ice for 7 min, and then ethanol precipitated and resuspended in 8 μL of H2O. S1 nuclease digestion was carried out on the resuspended DNA in a 10-μL reaction in 1X S1 buffer with 1 μL of S1 nuclease for 10 min at room temperature. The reaction was stopped by the addition of a Tris (pH 8)/ethylenediaminetetraacetic acid stop mix.
DNA for contour-clamped homogeneous electric field (CHEF) gel analysis was isolated in agarose plugs as described (J. L. Argueso, personal communication). CHEF gel analysis of yeast chromosomal DNAs was performed in 1% LE agarose gels with a switch time ramped from 47−170 sec at 165 volts for 66 hr in 0.5X TBE at 14° using a BioRad DRII electrophoresis chamber. Southern blots of CHEF gels were probed sequentially with a CEN2 probe and then with a SUL1 probe. The ratio of SUL1 to CEN2 hybridization was used to quantify the number of SUL1 repeats on each unique version of chromosome II, setting the ratio to 1.0 for chromosomes at the beginning of the experiment. In each generation sampled, only chromosome IIs that were present at >20% of the total were quantified. In the seven populations we detected 16 new versions of chromosome II that reached this cutoff for at least one sampling interval.
Polymerase chain reaction (PCR) and quantitative PCR (qPCR)
Smash-and-Grab DNA was used for qPCR as previously described (Di Rienzi et al. 2011) after being cleaned using the DNA Clean & Concentrator kit (Zymo Research). For each sample, the copy number of SUL1 was determined relative to the copy number of ACT1. The copy number of the locus for a given sample was normalized against the copy number of that locus in the original strain used to inoculate the ministat. A site was considered amplified if the copy number was ≥1.5.
Smash-and-Grab DNA was used for PCR amplification to obtain fragments used as probes and for Sanger sequencing. Primers are included in Table S3. PCR products of interest were purified with DNA Clean and Concentrator (Zymo Research) and primer extension sequencing was performed by GENEWIZ, Inc. (South Plainfield, NJ) using Applied Biosystems BigDye version 3.1. The reactions were run on Applied Biosystem’s 3730xl DNA Analyzer.
Clones and population array comparative genomic hybridization (aCGH) analysis
Frozen chemostat samples from generation ~200 were streaked onto limiting sulfate plates (medium as described previously plus 20g/L Difco agar). Single colonies were picked, and DNA was isolated by a modified Smash-and-Grab protocol (Hoffman and Winston 1987). For population analysis, DNA was extracted using the NIB-and-Grab method directly from the frozen sample. aCGH was performed using Agilent 4x44k microarrays with probes spaced every 290 nt on average. Hybridization was executed as described previously (Gresham et al. 2008). Microarray data from this article have been deposited in the Gene Expression Omnibus repository under accession GSE47854 (http://www.ncbi.nlm.nih.gov/geo/) and in the Princeton Microarray Database (http://puma.princeton.edu).
Competition experiment
The pairwise competition experiments were performed in ministats (Miller et al. 2013). Each competitor strain was cultured individually. Upon achieving steady state, the competitors were mixed in the indicated ratio. Each competition was conducted in two biological replicates for 15 generations after mixing. Samples were collected and analyzed twice daily. The proportion of GFP+ cells in the population was detected using a BD Accuri C6 flow cytometer (BD Biosciences). The data were plotted with ln[(dark cells/GFP+ cells)] vs. generations. The relative fitness coefficient was determined from the slope of the linear region by the use of linear regression analysis.
Nextera libraries and whole-genome sequencing
Genomic DNA libraries were prepared for Illumina sequencing using the Nextera sample preparation kit (Illumina). Barcoded libraries were quantified on an Invitrogen Qubit Fluorometer and submitted for 75 bp paired end sequencing on an Illumina HiSeq 2000. Read data have been deposited at the NCBI under BioSample accessions: SAMN02208069, SAMN02208070, SAMN02208071, SAMN02208072, SAMN02208073, SAMN02208074, and SAMN02208075. The reads were mapped against the genome sequence of the reference strain S288C (SacCer3) using mrsFAST (Hach et al. 2010). The sequence coverage of the nuclear genome ranged from 70 to 300x. Novel junctions and indels were identified with SplitReads (Karakoc et al. 2011). For single nucleotide variant (SNV) analysis, the reads were aligned with Burrows-Wheeler Aligner (Li and Durbin 2009) and SNVs were called via use of the Samtools (Li et al. 2009) mpileup command after applying standard filters (quality score <30). SNVs unique to the evolved clones were identified, annotated with a custom Python script (Pashkova et al. 2013), and further prioritized by manual examination with the Integrative Genomics Viewer (IGV) (Robinson et al. 2011).
Results
To visualize the diversity and dynamics of a population during 200 generations of adaptation to a sulfate-limiting environment, we first developed assays to establish clonal identity based on unique amplicons containing the SUL1 locus. We then took advantage of the specific and beneficial amplification of the SUL1 locus as an intrinsic marker of evolution to study and visualize clonal interference in evolving populations of yeast.
Adaptation to sulfate limitation selects for the amplification of SUL1 in S. cerevisiae
To characterize the evolutionary paths of yeast populations subjected to a constant selective pressure, in this case sulfate limitation, we performed seven parallel evolution experiments by using chemostat continuous cultures. Each experiment was initiated from a prototrophic haploid S. cerevisiae strain that had never before been exposed to long-term sulfate limitation. In sulfate-limiting conditions, the seven cultures reached steady-state growth with population sizes of ~109 cells. Six of the cultures were maintained in continuous growth for ~200 generations; the seventh culture (Pop1) was terminated early (at generation ~90) because the input media line became colonized. At the end of the experiment, a single clone from each population (six clones total) was analyzed for copy number variants and relative fitness.
As expected from previous studies (Gresham et al. 2008), we detected amplification of the SUL1 locus in each clone as assessed by aCGH (Figure 1, Figure 2A, and Figure S1). The number of SUL1 copies varied from three to five per haploid genome with the boundaries of the amplification differing in each individual clone. No other large structural changes were detected in any of the clones (Figure S1). Relative fitnesses of the six clones were estimated by competition with the wild-type ancestor in the same chemostat environment. All clones showed significantly higher fitness compared to the ancestral strain, with the relative fitness coefficients ranging from 36 to 53% (Table 1).
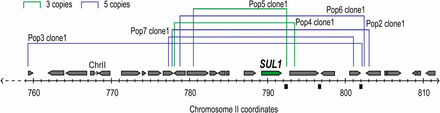
Unique SUL1 amplicons are observed in clones isolated from six evolution experiments. The map illustrates the location of SUL1, flanking open reading frames, and the origin of replication, ARS228. The lines above the map show the extent of the amplified segment observed by aCGH (Figure S1) for each clone: blue line, copy number 5; green line, copy number 3.
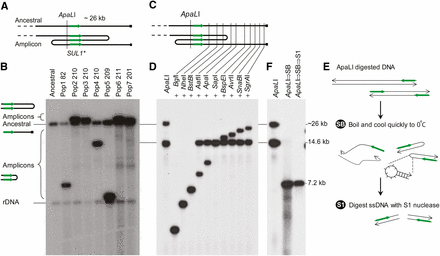
Analysis of SUL1 amplicons reveals inverted structures. (A) Map of the right telomeric region of chromosome II shows the position of SUL1, the relevant restriction enzyme sites, the deduced structure of Pop4 gen201 clone1, and the probe (SUL1*) used for Southern blot analysis. (B) Southern blot of ApaLI digests of seven clones containing SUL1 amplifications isolated from independent evolution experiments and hybridized with the SUL1 probe. The ancestral fragment corresponds to the telomere-proximal SUL1 fragment. The more prominent bands of variable sizes correspond to fragments with novel chromosomal junctions. (C) Indirect end-labeling of ApaLI double digests. The order of the lanes corresponds to the order in which the sites for the second restriction enzymes are found between ApaLI and the telomere. (D) Southern blot of the double digests. The series of bands of increasing sizes in the Southern blot indicates that the portion of the genome from SUL1 to the telomere is intact. Fragments that contain the amplicon junction comigrate with the expected fragments only up to the position of the junction. Second enzymes whose sites lie distal to the amplicon junction fail to make a second cleavage and produce the amplicon-specific ApaLI junction fragment. (E) A schematic illustrating the snap-back (SB)/S1 nuclease assay (S1). The rapid chilling of denatured ApaLI fragments only permits reformation of dsDNA if the molecule is self-complementary. S1 treatment degrades all single stranded fragments including the ssDNA in the loop. (F) Southern analysis of the snap-back/S1-nuclease assay of population 4 clone1 using the SUL1 probe. The 14.6-kb amplicon-specific ApaLI fragment generates an S1-resistant duplex molecule approximately half of its original size while the single strands of the ancestral fragment are degraded by S1 nuclease.
Fitness coefficient of evolved clones
| Population | Generations | Clones | Fitness Coefficient, % | SUL1 Copy Number |
|---|---|---|---|---|
| 2 | 210 | Clone1 | 43.33 ± 1.98 (n = 2) | 5 |
| 3 | 210 | Clone1 | 53.31 ± 0.88 (n = 2) | 5 |
| 4 | 210 | Clone1 | 37.69 ± 2.41 (n = 4) | 3 |
| 5 | 209 | Clone1 | 48.95 ± 0.03 (n = 2) | 3 |
| 6 | 211 | Clone1 | 46.81 ± 0.03 (n = 2) | 5 |
| 7 | 201 | Clone1 | 41.70 ± 5.94 (n = 2) | 5 |
| Population | Generations | Clones | Fitness Coefficient, % | SUL1 Copy Number |
|---|---|---|---|---|
| 2 | 210 | Clone1 | 43.33 ± 1.98 (n = 2) | 5 |
| 3 | 210 | Clone1 | 53.31 ± 0.88 (n = 2) | 5 |
| 4 | 210 | Clone1 | 37.69 ± 2.41 (n = 4) | 3 |
| 5 | 209 | Clone1 | 48.95 ± 0.03 (n = 2) | 3 |
| 6 | 211 | Clone1 | 46.81 ± 0.03 (n = 2) | 5 |
| 7 | 201 | Clone1 | 41.70 ± 5.94 (n = 2) | 5 |
| Population | Generations | Clones | Fitness Coefficient, % | SUL1 Copy Number |
|---|---|---|---|---|
| 2 | 210 | Clone1 | 43.33 ± 1.98 (n = 2) | 5 |
| 3 | 210 | Clone1 | 53.31 ± 0.88 (n = 2) | 5 |
| 4 | 210 | Clone1 | 37.69 ± 2.41 (n = 4) | 3 |
| 5 | 209 | Clone1 | 48.95 ± 0.03 (n = 2) | 3 |
| 6 | 211 | Clone1 | 46.81 ± 0.03 (n = 2) | 5 |
| 7 | 201 | Clone1 | 41.70 ± 5.94 (n = 2) | 5 |
| Population | Generations | Clones | Fitness Coefficient, % | SUL1 Copy Number |
|---|---|---|---|---|
| 2 | 210 | Clone1 | 43.33 ± 1.98 (n = 2) | 5 |
| 3 | 210 | Clone1 | 53.31 ± 0.88 (n = 2) | 5 |
| 4 | 210 | Clone1 | 37.69 ± 2.41 (n = 4) | 3 |
| 5 | 209 | Clone1 | 48.95 ± 0.03 (n = 2) | 3 |
| 6 | 211 | Clone1 | 46.81 ± 0.03 (n = 2) | 5 |
| 7 | 201 | Clone1 | 41.70 ± 5.94 (n = 2) | 5 |
The average fitness of clones with five copies of SUL1 (46.3%) did not significantly differ from that of clones with three copies of SUL1 (43.3%). This result contrasts with the significant difference in fitness between the ancestral strains carrying ~20 copies of a 2-μm plasmid with the SUL1 gene vs. one to two copies of a CEN plasmid with SUL1 (Table 2). There was also no significant correlation between the size of the amplicon and the relative fitness (data not shown). These data support the hypothesis that extra copies of SUL1, but not their absolute copy number above a minimal threshold nor the extent of flanking sequences, significantly affect the fitness of cells during growth under sulfate limitation. However, any additional mutations carried by these strains could confound our ability to detect such a trend.
Fitness associated with an increased copy number of SUL1
| Copy Number of SUL1 | Fitness Coefficient, % | Fitness Coefficient Corrected, % | |
|---|---|---|---|
| Empty CEN plasmid | 1 | −19 ± 1.41 (n = 2) | 0 |
| SUL1_CEN | 1−3 | 23 ± 4.93 (n = 3) | 42 |
| SUL1_2 micron | ~20 | 32 ± 3.21 (n = 3) | 51 |
| No plasmid | 1 | 0.03 ± 0.60 (n = 5) | − |
| Copy Number of SUL1 | Fitness Coefficient, % | Fitness Coefficient Corrected, % | |
|---|---|---|---|
| Empty CEN plasmid | 1 | −19 ± 1.41 (n = 2) | 0 |
| SUL1_CEN | 1−3 | 23 ± 4.93 (n = 3) | 42 |
| SUL1_2 micron | ~20 | 32 ± 3.21 (n = 3) | 51 |
| No plasmid | 1 | 0.03 ± 0.60 (n = 5) | − |
| Copy Number of SUL1 | Fitness Coefficient, % | Fitness Coefficient Corrected, % | |
|---|---|---|---|
| Empty CEN plasmid | 1 | −19 ± 1.41 (n = 2) | 0 |
| SUL1_CEN | 1−3 | 23 ± 4.93 (n = 3) | 42 |
| SUL1_2 micron | ~20 | 32 ± 3.21 (n = 3) | 51 |
| No plasmid | 1 | 0.03 ± 0.60 (n = 5) | − |
| Copy Number of SUL1 | Fitness Coefficient, % | Fitness Coefficient Corrected, % | |
|---|---|---|---|
| Empty CEN plasmid | 1 | −19 ± 1.41 (n = 2) | 0 |
| SUL1_CEN | 1−3 | 23 ± 4.93 (n = 3) | 42 |
| SUL1_2 micron | ~20 | 32 ± 3.21 (n = 3) | 51 |
| No plasmid | 1 | 0.03 ± 0.60 (n = 5) | − |
SUL1 amplicons have an inverted repeat structure
To determine the chromosomal location of the additional copies of SUL1 in each clone, we performed CHEF gel analysis coupled with Southern blot hybridization using SUL1 and CEN2 probes (Figure S2). In each case, chromosome II migrated more slowly and migrated the same distance as the band hybridized by the SUL1 probe (data not shown), consistent with the amplified SUL1 sequences residing on chromosome II. Because sequence analysis of a previously characterized SUL1 amplicon revealed a tandem inverted structure for the additional copies of SUL1 (Araya et al. 2010), we devised electrophoretic tests to detect potential inverted structures. These approaches allow both qualitative and quantitative characterization of clonal amplicons at the SUL1 locus. DNA from the ancestral strain was digested with ApaL1, a Southern blot was hybridized with a SUL1 probe, and the expected band of approximately 26 kb was observed (Figure 2B). Although this fragment also was detected in the amplified clones, an additional, variable fragment was detected as well (Figure 2B). The size of this additional band was roughly consistent with the aCGH data, assuming an inverted repeat orientation; the size of the amplification-specific band was equal to twice the distance from the ApaLI site to the telomere-proximal amplicon junction (Figure 2C). Similar results were found for the centromere-proximal junction using EcoNI digestion (data not shown).
To confirm the inverted structure, we conducted indirect end-labeling using SUL1 as a probe on Southern blots of genomic DNA cleaved with ApaLI and a series of second enzymes that cut at increasing distances from SUL1 toward the telomere (Figure 2, C and D). The doubly digested DNA from the ancestral strain produces a ladder of fragments of increasing size reflecting the order of restriction sites in the ancestral genome. Evolved clones with an inverted triplication, such as illustrated in Figure 2A, produce the same ladder of fragments because the telomere-proximal copy of SUL1 is identical to that found in the ancestral strain. However, the ApaLI fragment unique to the inversion junction can only be cleaved by the enzymes that recognize the more centromere-proximal sites. All enzymes that recognize sites beyond the amplification junction leave the amplicon-specific ApaLI fragment intact (Pop4 210 clone1, Figure 2D). Indirect end labeling using EcoNI (Figure S3) allowed us to map the inversion junction on the centromere-proximal side of SUL1 for many of the clones. We analyzed each of the six clones in an identical manner and obtained results that are consistent with inversion junctions at the boundaries of the amplifications we found using aCGH (data not shown). As a final test of the inverted structure of the amplicons, we carried out snap-back assays (Tanaka et al. 2005). If the amplicons-specific ApaLI fragments were inverted, then denaturation and rapid cooling would produce duplex hairpin fragments that are resistant to digestion with S1-nuclease (Figure 2E). Each of the six clones had an S1-resistant snapback DNA fragment of a size that is approximately half of the original ApaLI fragment (Figure 2F and data not shown). Snap-back analyses of the centromere-proximal junctions confirmed the inverted structure of the amplicons (Figure S3 and data not shown).
Sequencing of the SUL1 amplicon junctions reveals inverted microhomologies
To map in detail the SUL1 amplicon junctions of six of the evolved clones isolated at generation 200, we applied a split-read sequencing method designed to identify the exact junctions for complex events (tandem duplication, inversion and deletion) (Karakoc et al. 2011; Figure 3A). The accumulation of balanced split-reads (i.e., split in the middle of the read) and unbalanced split-reads (i.e., split on one side of the read) at a specific genomic locus is the signature of a rearrangement junction. For population 4 clone 1, we identified 17 split-reads mapping within the 893 bp windows encompassing the left junction and 18 split-reads within the 507 bp window for the right junctions identified by aCGH (Figure 3, B and C and Table 3). All but one of the junctions showed pairs of 5- to 10-bp interrupted palindromic sequences flanking the junction; the twelfth junction was located in a CAG repeat region (Table 3). In confirmation of the aCGH data, the 12 junctions occurred at unique sites. The median distance between the two halves of the interrupted palindromic sequences was 40 bp (Table 3). The orientations of the split reads also confirmed the inverted structure of the amplicons. The interrupted palindromes are similar in structure to the junction sequences of a previously analyzed, evolved clone with a SUL1 amplicon bearing an inverted repeat structure (Araya et al. 2010).
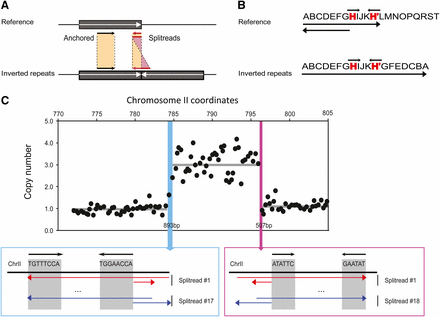
Detection of unique junctions using split-read methods. (A) Schematic diagram for the mapping of paired-end sequences at the junction. In a pair of reads where only one read is mapped (anchored), the second unmapped read is split into two parts and mapped to the genome. (B) Diagram for the split-read at the junction that contains an interrupted inverted repeat (H). (C) Expanded view of the last 35 kb of chromosome II containing the amplification of the SUL1 locus for Pop4 210 clone 1. The blue and pink boxes correspond to the regions in which the junctions of the amplification have occurred. The accumulation of split-reads that include the short inverted repeats (black arrows) indicates the junctions of the rearrangements in the evolved genome.
Junction signatures
| Strains | Left Junction | Right Junction | ||||||
|---|---|---|---|---|---|---|---|---|
| Elements | Palindromes | No. Reads | Loop, nt | Elements | Palindromes | No. Reads | Loop, nt | |
| 2_210 Clone1 | YBR287w | GCCATT-AATGGC | 24 | 44 | MAL31 | GGTGC-GCACC | 22 | 40 |
| 3_210 Clone1 | PPS1 | CATCAT-ATGAGG | 21 | 225 | Intergenic | GTTTTTTCA-TGAAAAAAC | 30 | 22 |
| 4_210 Clone1 | CTP1 | TGTTTCCA-TGGAACCA | 17 | 27 | PCA1 | ATATTC-GAATAT | 18 | 29 |
| 5_209 Clone1 | SNF5 | CAG repeats | ARS228 | ATGAATCT-AGAT_CAT | 35 | 99 | ||
| 6_211 Clone1 | APM3 | TTCCATGGA- TCCAGGGAA | 25 | 114 | Intergenic | GTTTTTTCA- TGAAAAAAC | 15 | 22 |
| 7_201 Clone1 | SNF5-APE3 | ACTTGACCAA-TTGGTCAAGT | 28 | 4180 | MAL33 | TACCAATG-CATTGGTA | 32 | 22 |
| Strains | Left Junction | Right Junction | ||||||
|---|---|---|---|---|---|---|---|---|
| Elements | Palindromes | No. Reads | Loop, nt | Elements | Palindromes | No. Reads | Loop, nt | |
| 2_210 Clone1 | YBR287w | GCCATT-AATGGC | 24 | 44 | MAL31 | GGTGC-GCACC | 22 | 40 |
| 3_210 Clone1 | PPS1 | CATCAT-ATGAGG | 21 | 225 | Intergenic | GTTTTTTCA-TGAAAAAAC | 30 | 22 |
| 4_210 Clone1 | CTP1 | TGTTTCCA-TGGAACCA | 17 | 27 | PCA1 | ATATTC-GAATAT | 18 | 29 |
| 5_209 Clone1 | SNF5 | CAG repeats | ARS228 | ATGAATCT-AGAT_CAT | 35 | 99 | ||
| 6_211 Clone1 | APM3 | TTCCATGGA- TCCAGGGAA | 25 | 114 | Intergenic | GTTTTTTCA- TGAAAAAAC | 15 | 22 |
| 7_201 Clone1 | SNF5-APE3 | ACTTGACCAA-TTGGTCAAGT | 28 | 4180 | MAL33 | TACCAATG-CATTGGTA | 32 | 22 |
Bold indicates imperfect palindromic nucleotides.
| Strains | Left Junction | Right Junction | ||||||
|---|---|---|---|---|---|---|---|---|
| Elements | Palindromes | No. Reads | Loop, nt | Elements | Palindromes | No. Reads | Loop, nt | |
| 2_210 Clone1 | YBR287w | GCCATT-AATGGC | 24 | 44 | MAL31 | GGTGC-GCACC | 22 | 40 |
| 3_210 Clone1 | PPS1 | CATCAT-ATGAGG | 21 | 225 | Intergenic | GTTTTTTCA-TGAAAAAAC | 30 | 22 |
| 4_210 Clone1 | CTP1 | TGTTTCCA-TGGAACCA | 17 | 27 | PCA1 | ATATTC-GAATAT | 18 | 29 |
| 5_209 Clone1 | SNF5 | CAG repeats | ARS228 | ATGAATCT-AGAT_CAT | 35 | 99 | ||
| 6_211 Clone1 | APM3 | TTCCATGGA- TCCAGGGAA | 25 | 114 | Intergenic | GTTTTTTCA- TGAAAAAAC | 15 | 22 |
| 7_201 Clone1 | SNF5-APE3 | ACTTGACCAA-TTGGTCAAGT | 28 | 4180 | MAL33 | TACCAATG-CATTGGTA | 32 | 22 |
| Strains | Left Junction | Right Junction | ||||||
|---|---|---|---|---|---|---|---|---|
| Elements | Palindromes | No. Reads | Loop, nt | Elements | Palindromes | No. Reads | Loop, nt | |
| 2_210 Clone1 | YBR287w | GCCATT-AATGGC | 24 | 44 | MAL31 | GGTGC-GCACC | 22 | 40 |
| 3_210 Clone1 | PPS1 | CATCAT-ATGAGG | 21 | 225 | Intergenic | GTTTTTTCA-TGAAAAAAC | 30 | 22 |
| 4_210 Clone1 | CTP1 | TGTTTCCA-TGGAACCA | 17 | 27 | PCA1 | ATATTC-GAATAT | 18 | 29 |
| 5_209 Clone1 | SNF5 | CAG repeats | ARS228 | ATGAATCT-AGAT_CAT | 35 | 99 | ||
| 6_211 Clone1 | APM3 | TTCCATGGA- TCCAGGGAA | 25 | 114 | Intergenic | GTTTTTTCA- TGAAAAAAC | 15 | 22 |
| 7_201 Clone1 | SNF5-APE3 | ACTTGACCAA-TTGGTCAAGT | 28 | 4180 | MAL33 | TACCAATG-CATTGGTA | 32 | 22 |
Bold indicates imperfect palindromic nucleotides.
SUL1 amplification occurs early during the adaptation to sulfate limitation
To better understand the evolutionary dynamics of SUL1 amplification, we determined when amplicons appeared during the 200 generations of adaptation to sulfate limitation. We used real-time, qPCR on genomic DNA collected from the evolving populations at roughly 50-generation intervals (Figure 4). By generation ~50, we detected amplification of SUL1 in approximately half of the populations, and by generation ~100, in all of the populations. The average population copy number of SUL1 in the six completed evolution experiments ranged from 1.9 to 4.2, values consistent with the estimates of copy number in the final clones. Although these results confirm that adaptation to sulfate limitation proceeds via the amplification of the SUL1 gene, the kinetics of the amplification and the final copy number achieved varied between the replicate evolution experiments, suggesting that each population experienced different evolutionary trajectories over the course of sulfate-limited adaptation.
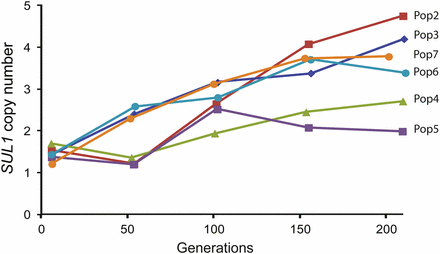
Evolutionary dynamics of SUL1 amplification of experimental populations evolved in sulfate limitation medium. The copy number of SUL1 was assessed using qPCR analysis on samples taken from Populations 2 through 7 every ~50 generations.
Clonal interference is commonly observed during the adaptation to sulfate limitation
Population-level data from experimental evolution can be analyzed for evidence of clonal interference, suggesting the origin and fate of adaptive mutations. Because the qPCR analysis provides only population averages of the SUL1 amplification, we performed electrophoresis-based analysis of SUL1 amplicons to track the frequencies of distinct subpopulations. We isolated DNA from chemostat samples at regular intervals over each ∼200 generation experiment and digested aliquots separately with EcoN1 and ApaL1 (Figure 5, A−C). Using SUL1 as the hybridization probe, we were able to detect when new amplification junctions arose (Figure 5, B and C). Hybridization of the Southern blots with a probe from a genomic region with a copy number of one (ARS305; Figure 5, B and C) allowed us to quantify the prevalence of each amplicon junction over the course of the sulfate-limited growth. With this assay, we were able not only to follow the overall dynamics of the SUL1 amplification but also to identify subpopulations that carry unique SUL1 amplification junctions and to track their frequencies in the population. Although we can assess the relative abundance of each amplicon junction (Figure 5D), it should be noted that the relative abundance of each subpopulation reported by this assay is necessarily a composite of the subpopulation frequency and the clonal copy number of the SUL1 amplicon. To disentangle these two variables, we simultaneously isolated genomic DNA in agarose plugs for karyotype analysis using CHEF gel electrophoresis (Figure 5D). Using CEN2 and SUL1 as hybridization probes, we could detect when increases in the size of chromosome II occurred (Figure 5E) and the number of copies of SUL1 on each new version of chromosome II (Figure 5F). During the course of the seven evolution experiments, we detected a minimum of 16 new versions of chromosome II with chromosomes containing three copies of SUL1 being replaced by chromosomes with higher copy numbers over time. Although we could not measure significant differences in fitness for three vs. five copies of SUL1 (43.3% vs. 46.3%, respectively), five of the six evolution experiments that reached ~200 generation were being overtaken by higher copy-number clones. Most of the increases in copy number were not accompanied by changes in junction fragments, suggesting that the increase in copy number was a consequence of unequal crossing over that expanded SUL1 arrays from three to five copies.
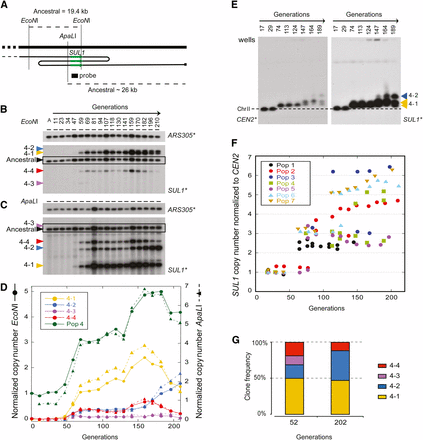
Kinetics of SUL1 amplicon formation during ~200 generations of sulfate-limited growth. (A) A map of the SUL1 region of chromosome II showing the positions of ApaLI and EcoNI restriction sites used to digest DNA isolated from different samples of evolution #4 (Pop4). The location of SUL1, a hypothetical structure of an inverted amplicon, and the probe used for Southern hybridization are also shown. (B) EcoNI digestion and electrophoretic separation of chromosomal fragments recovered during sulfate limited growth. Top, control hybridization of the Southern blot with a single copy sequence ARS305. Bottom, hybridization of the EcoNI blot with SUL1. (C) ApaLI digestion and electrophoretic separation of the same DNA samples as in panel B. Top, hybridization of the blot with ARS305. Bottom, hybridization of the blot with SUL1. (D). Quantification of different amplicons during evolution #4 (Pop4) using ARS305 hybridization for normalization. A minimum of four unique amplicons were detected for both digests. Their pattern of abundance, appearance and disappearance identifies which proximal and distal junction fragments make up individual amplicons (for example, 4-2). (E) CHEF gel analysis of population samples of Pop4. The Southern blot was hybridized sequentially with a CEN2 probe (left) and then a SUL1 probe (right). (F) Determination of SUL1 copy number on variant copies of chromosome II. For each form of chromosome II that accounted for at least 20% of the chromosome IIs in the population, the ratio of SUL1/CEN2 was normalized to the ratio at the start of the evolution experiment to determine the copy number of SUL1 for each variant chromosome II. For several evolution experiments multiple different forms of chromosome II transiently coexisted. (G) Chart showing the proportion of clones from Pop4 detected by single clone analysis using ApaL1 digestion.
Using these two gel assays, we detected the first SUL1 amplicons between 46 and 71 generations (Pop4, Figure 5, B−E and Pop1−3, 5−7, Figure S4). Over the course of all evolution experiments, we observed the presence of multiple subpopulations, each carrying different SUL1 amplicons. The subpopulations in all of the evolution experiments demonstrated two distinct behaviors: at least one subpopulation persisted throughout the course of the experiment, and additional transient subpopulations rose to different frequencies and then fell below the level of detection before the end of the experiment.
In Population 4, several subpopulations were already observable at generation 59, and most persisted throughout the course of the evolution experiment although they fluctuated in frequency over time (Figure 5B−D). For example, after generation 150, the two predominant subpopulations (4-1 and 4-4) declined in frequency whereas the third subpopulation expanded (4-2; Figure 5E)—a result consistent with clonal interference. These data clearly demonstrate the presence of multiple adaptive events and reflect a greater diversity within subpopulations.
To further disambiguate clone frequency and SUL1 copy number, we verified these results by examining 45 clones from Population 4 at generations 52 and 202 by using the electrophoretic assay focused on the centromere-proximal junction of the SUL1 amplicon. The increase in frequency of clones corresponding to subpopulation 4-2 at the expense of subpopulation 4-3 between generations 50 and 200 matches our observations from the population analysis (Figure 5F).
Because all strains generate wild-type ApaLI and EcoNI bands in the electrophoretic assay, we were unable to determine what fraction of the populations did not carry an amplicon, or whether the SUL1 amplification had become fixed. To specifically investigate the dynamics of SUL1 copy number and population frequency of its amplification throughout the course of each evolution experiment, we used qPCR on a total of 506 independent clones, isolated from each experiment at generations ~50 and ~200 (Table S1). At generation ~50 we did not detect any amplification of SUL1 in two of the populations (Population 2 and Population 5) whereas in the four remaining populations, 21–97% of clones contained a SUL1 amplification event. At generation ~200, SUL1 amplification was present at high frequency in four of the six populations (80–98% of clones) and had apparently become fixed in two populations (Pop 2 and 7; Figure 6). Some of the clones examined may have lost their amplicon by homologous recombination during the period of nonselective growth after removal from the chemostat. However, the CHEF analysis of population samples not subjected to nonselective growth (Figure 5E and Figure S4) confirmed that in some of the evolution experiments, a small subset of cells (from 1 to 15%) still retained the ancestral-sized chromosome II. In general, the presence or absence of the SUL1 amplification at generation ~50 could not be used as a predictor for fixation at generation ~200. These data establish the fixation of SUL1 amplification at ~200 generations in two populations (Population 2 and 7). Even in these two populations, multiple coexisting subpopulations arose together possibly indicating the presence of other mutations in these populations as well.
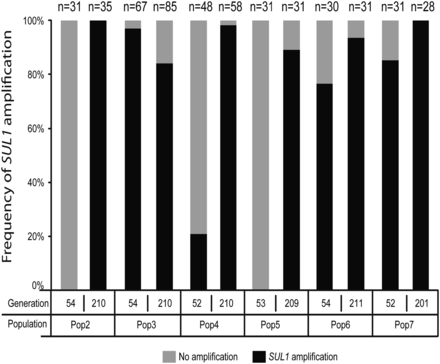
Frequency of SUL1 amplification at generations ~50 and ~200. qPCR was used to determine the percentage of clones with SUL1 amplification at generations ~50 and ~200 in six populations (in black) vs. percentage of clones found with only one copy of SUL1 (in gray).
Alternative adaptive trajectories are rarely observed in populations evolving under sulfate limitation
The varying dynamics of amplification observed among different subpopulations, the decrease in the frequency of SUL1 amplification in Population 3, and the fact that the SUL1 amplification is not fixed in most of the populations at generation ~200 suggest that additional mutations have occurred that may interfere with the fixation of SUL1 amplifications (Figure 6). We looked directly for such possible mutations by isolating nine clones from Population 3 that did not contain a SUL1 amplicon at generation ~200 and determining their relative fitness. Except for one clone, the fitness change compared with the ancestral strain was minimal (Table 4). One clone was 26.25% more fit than the ancestral strain. To rule out the possibility that cells with SUL1 amplification were selected during the 15 generations of competition used to determine the relative fitness, we performed qPCR on a sample from the last day of the competition experiment in which 96% of the cells corresponded to the Population 3 clone. Amplification at the SUL1 locus was not detected by qPCR, and the absence of SUL1 amplification or other major variations was confirmed by aCGH (Table 4 and Figure S5).
Fitness coefficient of clones from Pop3 at generation 210 without SUL1 amplification
| Clones | Fitness Coefficient, % | SUL1 Copy Number |
|---|---|---|
| 2 | −1.25 ± 0.77 (n = 2) | 1 |
| 3 | −2.59 ± 2.92 (n = 2) | 1 |
| 4 | 0.31 ± 4.81 (n = 2) | 1 |
| 5 | −2.12 ± 1.98 (n = 2) | 1 |
| 6 | 0.89 ± 1.36 (n = 2) | 1 |
| 7 | 0.33 ± 0.77 (n = 2) | 1 |
| 8 | 1.58 ± 2.40 (n = 2) | 1 |
| 9 | −2.88 ± 0.095 (n = 2) | 1 |
| 10 | 26.25 ± 2.68 (n = 4) | 1 |
| Clones | Fitness Coefficient, % | SUL1 Copy Number |
|---|---|---|
| 2 | −1.25 ± 0.77 (n = 2) | 1 |
| 3 | −2.59 ± 2.92 (n = 2) | 1 |
| 4 | 0.31 ± 4.81 (n = 2) | 1 |
| 5 | −2.12 ± 1.98 (n = 2) | 1 |
| 6 | 0.89 ± 1.36 (n = 2) | 1 |
| 7 | 0.33 ± 0.77 (n = 2) | 1 |
| 8 | 1.58 ± 2.40 (n = 2) | 1 |
| 9 | −2.88 ± 0.095 (n = 2) | 1 |
| 10 | 26.25 ± 2.68 (n = 4) | 1 |
| Clones | Fitness Coefficient, % | SUL1 Copy Number |
|---|---|---|
| 2 | −1.25 ± 0.77 (n = 2) | 1 |
| 3 | −2.59 ± 2.92 (n = 2) | 1 |
| 4 | 0.31 ± 4.81 (n = 2) | 1 |
| 5 | −2.12 ± 1.98 (n = 2) | 1 |
| 6 | 0.89 ± 1.36 (n = 2) | 1 |
| 7 | 0.33 ± 0.77 (n = 2) | 1 |
| 8 | 1.58 ± 2.40 (n = 2) | 1 |
| 9 | −2.88 ± 0.095 (n = 2) | 1 |
| 10 | 26.25 ± 2.68 (n = 4) | 1 |
| Clones | Fitness Coefficient, % | SUL1 Copy Number |
|---|---|---|
| 2 | −1.25 ± 0.77 (n = 2) | 1 |
| 3 | −2.59 ± 2.92 (n = 2) | 1 |
| 4 | 0.31 ± 4.81 (n = 2) | 1 |
| 5 | −2.12 ± 1.98 (n = 2) | 1 |
| 6 | 0.89 ± 1.36 (n = 2) | 1 |
| 7 | 0.33 ± 0.77 (n = 2) | 1 |
| 8 | 1.58 ± 2.40 (n = 2) | 1 |
| 9 | −2.88 ± 0.095 (n = 2) | 1 |
| 10 | 26.25 ± 2.68 (n = 4) | 1 |
To investigate the genetic changes underlying the fitness increase of this clone, we sequenced its genome at 60x coverage and detected de novo mutations relative to the ancestral strain. Consistent with the aCGH data, no large CNVs were detected in this strain. Our initial analysis predicted a total of three point mutations: one in a telomeric region and two nonsynonymous mutations in coding regions (Table S2). We validated the two nonsynonymous mutation calls by Sanger sequencing. No mutations were predicted in the coding sequence or the upstream regulatory sequences of SUL1, ruling out an increase in the expression of SUL1 due to a mutation in cis. As a convergent mutation may provide additional evidence of adaptive events, we compared this list of mutations to the genome sequences obtained for the additional evolved clones, plus those we previously detected by tiling arrays (Gresham et al. 2008) and by whole-genome sequencing (Araya et al. 2010). We found that SGF73 was mutated in two clones from this study (Population 3 clone described previously and a final clone from Population 7) and two other clones from our previous studies (Gresham et al. 2008; Araya et al. 2010). Sgf73 is a subunit of the SAGA histone acetylase complex required for the assembly of the histone deubiquitination module and the yeast ortholog of Ataxin-7 (Kohler et al. 2008). We confirmed the mutations in SGF73 in these four clones by Sanger sequencing (Figure S6). In every instance, the mutation is a nonsense mutation predicted to truncate the SGF73 gene product, suggesting that inactivation of SGF73 may be advantageous in sulfate limitation. Interestingly, two of the evolved clones with SGF73 mutations also carried the SUL1 amplification while two did not. To examine further the physiological effect of this allele, we determined the relative fitness in both sulfate and glucose limitation of a strain deleted for SGF73 [obtained from the Yeast deletion collection (Giaever et al. 2002)]. This strain had never previously been cultivated in sulfate-limited media. We found that the deletion of SGF73 has a small deleterious effect in glucose limitation (−3.64% ± 0.62) but has an increased fitness of 24.3% in sulfate limitation, demonstrating that the positive fitness effects of this mutation may be specific to sulfate limitation. No previous connection to sulfate metabolism has been reported. Although the role that this gene plays in a sulfate-limited environment has yet to be elucidated, its positive effect on fitness under sulfate-limiting conditions strongly suggests this mutation offers an alternative adaptive trajectory to this strong selective pressure.
Discussion
The adaptation of S. cerevisiae to limited sulfate conditions during long-term evolution experiments provides a powerful approach to study an adaptive trajectory. In this work, we show that the vast majority of SUL1 amplification products that arise in sulfate-limited growth are in situ inverted amplicons with unique junctions coinciding with genomic sequences that consist of short, interrupted palindromes, a result we first discovered for a single clone (Araya et al. 2010). The unique structure of these amplicons and their predictable occurrence during sulfate-limited growth provides a rare opportunity to study potential mechanisms that generate this interstitial, inverted form of gene amplification (Brewer et al. 2011). This system also provides a unique opportunity to study how gene amplification contributes to adaptation under strong selection, a topic of much recent interest (Sonti and Roth 1989; Koszul et al. 2004; Gresham et al. 2008, 2010; Kugelberg et al. 2010; Blount et al. 2012). By following the trajectory of amplification of SUL1 over time with qPCR and electrophoretic techniques, we find that genetically distinct subpopulations arise within ~60 generations, coexist to variable degrees over ~200 generations, and change in relative representation as one adaptive variant replaces another. Thus, these evolution experiments display direct support for clonal interference. Unlike point mutation, amplification as an adaptive strategy has the unique property that it is easily reversible by intrachromosomal homologous recombination (Andersson et al. 1998). Further analysis of the fitness and rates of amplification and contraction at this locus across conditions will be required to resolve the importance of this possibility.
Although we previously demonstrated that strains with increased SUL1 copy number also showed increased RNA abundance (Gresham et al. 2008), we have not measured RNA content for the particular populations reported here. Quantitative assays of SUL1 RNA and protein levels would allow us to measure the extent of correlation between copy number, mRNA and protein levels, and let us directly measure how these molecular phenotypes correlate with fitness in sulfate limitation and other conditions.
Among rare clones from the last day of Population 3 that had either persisted without SUL1 amplification or had recently lost their SUL1 amplicons, we found one clone that had acquired an adaptive point mutation in the SGF73 gene. The fitness increase of the SGF73 mutation was substantially lower than that provided by SUL1 amplification (25% vs. 43%), suggesting that adaptive point mutations, if and when they occur, cannot compete with the added advantage that SUL1 amplification provides. In a previous study, we found that SGF73 mutations rose to 15–20% allele frequency in two populations; however, the SUL1 amplification status of these subpopulations was not determined (Gresham et al. 2008).
The observation of subpopulations that are genetically distinct at one locus throughout the course of the evolution experiments is consistent with previous studies in large populations of bacteria that demonstrate periodic selection of more fit clones harboring different beneficial mutations in the same gene (Notley-McRobb and Ferenci 2000; Lee and Marx 2013). The coexistence of transient large-scale rearrangements at the SUL1 locus in all seven of the evolution experiments provides direct evidence of strong competition among the evolving subpopulations. Although SUL1 amplification was expected in each of the populations, the apparent simultaneous occurrence of multiple independent amplicons within each single culture had not been systematically observed in previous studies. With a starting population size in the chemostats of 109 individuals and an estimated rate of amplicon formation of 10−7/cell/division (Payen et al. 2008), we considered the possibility that amplicons may have pre-existed in the inoculum cultures, and that they swept the population, as predicted from theoretical models (Wahl and Krakauer 2000). This possibility may explain why we first detected amplicons at roughly the same time in different chemostat cultures. This finding could also mean that there is a low probability that an advantageous mutation can occur de novo after the chemostat culture has been established or, if it does arise, that it fails to sweep the population.
Ultra-deep sequencing of the SUL1 flanking sequences from the ancestral strain inoculum and from populations over the course of the evolution experiment might allow us to detect rare initial and failed de-novo events. In addition to the common form of amplification event, we also note another trend over the course of the evolution experiments: the major amplicon in five of the seven populations increased in copy number from three to five as judged by the jumps in chromosome II size (Figure 5, D and E and Figure S4).
Recently, Yona et al. (2012) have proposed that large copy-number variants are first acquired during the course of an evolution experiment and are rapidly replaced by a more refined adaptive solution (e.g., elevated expression of few genes). In our case, we anticipated the appearance of point mutations that would increase SUL1 expression. The sequencing data of our clones did not reveal any mutations within the promoter of SUL1 and a previous study likewise did not show any increase in SUL1 expression independent of the effect of the copy number in clones isolated at 120−300 generations (Gresham et al. 2008). Because even small regions of aneuploidy will cause an overproduction of proteins that could lead to an accumulation of misfolded proteins, a condition known as proteotoxic stress (reviewed in Tang and Amon 2013), we imagined that there would be a fitness cost for retaining large stretches of SUL1 flanking DNA as part of the amplicon. As a consequence, we expected to see that the shortened amplicons would replace larger amplicons over time. However, that was not the case: for example, clone 4-4 (Figure 5, B and C) contained the smallest amplicons of the four coexisting subpopulations and was on its way to extinction as other larger amplicons remained. Possible explanations for this result are that additional driver genes might be present on the longer amplicons, or beneficial point mutations could have arisen in the genome. As discussed previously, we detected such a beneficial point mutation in the SGF73 gene in a clone that appeared to have escaped or lost its SUL1 amplicon. Independent mutations in this gene were found in other cultures, most notably in a final clone from Population 7 that contained both SUL1 amplification and the nonsense mutation of SGF73. The four independent mutations leading to the truncation of Sgf73, are an example of convergent evolution at the gene level (Woods et al. 2006; Tenaillon et al. 2012). Although we did not test it directly, it appears that the fitnesses associated with these two types of events are not additive, as the fitness of this particular clone is actually below the average of the six tested clones. Because our experiment was limited to ~200 generations, we cannot rule out the possibility that the beneficial mutations acquired and observed in our study are transient and that they would have been replaced by a more efficient and sustainable solution given more time. Exploring the stability and the persistence of the SUL1 amplification over a longer period of time will be key to understanding the dynamics between transient and possibly costly aneuploidy events and more refined mechanisms of adaptation to nutrient stress.
Our evolutionary studies in sulfate-limited chemostats also provide an efficient experimental tool to explore the mechanism that produces inverted, in situ amplicons. Because SUL1 is located near the telomere of chromosome II, the intrinsic instability associated with subtelomeric genomic domains may play a pivotal role in amplification of this region. However, the predominant form of instability associated with subtelomeres has been ascribed to their high levels of inter- and intrachromosomal recombination (Pryde and Louis 1997). Little is known about the instability of such inverted repeat structures; however, repeat numbers of three or five could easily resolve to single copy by unequal recombination or pop-out recombination at the alternating, directly repeating copies of the amplicon. A previous study reported that the frequency of loss of direct tandem duplications is positively correlated with the size of the amplicon (Koszul et al. 2006). The possible role of recombination in the generation of these palindrome-associated, inverted amplification events remains obscure. We favor an alternative mechanism for the generation of these specific inverted amplicons based on aberrant replication fork processing (Brewer et al. 2011). Because we have demonstrated that inverted SUL1 amplicons are the predominant solution to growth in sulfate-limited chemostats, we are now in position to dissect the genetic and molecular requirements for this under-explored and under-appreciated mode of gene amplification.
Acknowledgments
We thank the members of the Brewer/Raghuraman and Dunham labs for helpful discussions and Stan Fields, Gilles Fischer, and Ben Kerr for helpful comments on the manuscript. We also thank David Breslow and Jonathan Weissman for their eGFP construct, Barry Dion for constructing the ho::KanMX-GFP strain used for our competition experiments, and Angelika Amon, Eduardo Torres, and the Nickerson lab for assistance with the DNA sequencing. This work was supported by NSF grant 1120425 and NIGMS grants GM18926 to BJB and MKR and GM094306 to MJD. MJD is a Rita Allen Foundation Scholar and a CIFAR Fellow. ABS was supported by T32 AG000057 and F30CA165440.
Footnotes
Communicating editor: B. J. Andrews
Literature Cited
Author notes
Supporting information is available online at http://www.g3journal.org/lookup/suppl/doi:10.1534/g3.113.009365/-/DC1.
Microarray data from this article have been deposited in the Gene expression Omnibus (GEO) repository under accession GSE47854 (http://www.ncbi.nlm.nih.gov/geo/) and in the Princeton Microarray Database (http://puma.princeton.edu).
Sequencing data have been deposited with the National Center for Biotechnology Information under BioSample accessions SAMN02208069, SAMN02208070, SAMN02208071, SAMN02208072, SAMN02208073, SAMN02208074, and SAMN02208075.
Present address: Department of Microbiology, Cornell University, Ithaca, NY 14853.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}