





Abstract
The germ line efficiently combats numerous genotoxic insults to ensure the high fidelity propagation of unaltered genomic information across generations. Yet, germ cells in most metazoans also intentionally create double-strand breaks (DSBs) to promote DNA exchange between parental chromosomes, a process known as crossing over. Homologous recombination is employed in the repair of both genotoxic lesions and programmed DSBs, and many of the core DNA repair proteins function in both processes. In addition, DNA repair efficiency and crossover (CO) distribution are both influenced by local and global differences in chromatin structure, yet the interplay between chromatin structure, genome integrity, and meiotic fidelity is still poorly understood. We have used the xnd-1 mutant of Caenorhabditis elegans to explore the relationship between genome integrity and crossover formation. Known for its role in ensuring X chromosome CO formation and germ line development, we show that xnd-1 also regulates genome stability. xnd-1 mutants exhibited a mortal germ line, high embryonic lethality, high incidence of males, and sensitivity to ionizing radiation. We discovered that a hypomorphic allele of mys-1 suppressed these genome instability phenotypes of xnd-1, but did not suppress the CO defects, suggesting it serves as a separation-of-function allele. mys-1 encodes a histone acetyltransferase, whose homolog Tip60 acetylates H2AK5, a histone mark associated with transcriptional activation that is increased in xnd-1 mutant germ lines, raising the possibility that thresholds of H2AK5ac may differentially influence distinct germ line repair events. We also show that xnd-1 regulated him-5 transcriptionally, independently of mys-1, and that ectopic expression of him-5 suppressed the CO defects of xnd-1. Our work provides xnd-1 as a model in which to study the link between chromatin factors, gene expression, and genome stability.
Genome stability encompasses the mechanisms that ensure the integrity of DNA amid constant insults, the most toxic of which are DNA double-strand breaks (DSBs). DSBs emanate from both endogenous sources (replication stress and nucleases) and exogenous sources [e.g., ionizing radiation (IR)]. Induction of DSBs triggers the DNA damage response (DDR), a collection of systems that sense, respond to, and repair damaged DNA. Part of the DDR includes the initiation of cell cycle arrest to promote DNA repair, or the initiation of apoptosis if the damage cannot be repaired.
Despite their toxicity, the formation and repair of DSBs in the germ line is essential for the establishment of crossovers (COs) between homologous chromosomes during meiosis I. DSBs are purposefully created as the first step in meiotic CO formation by the topoisomerase II superfamily member Spo11 (Keeney et al. 1997; Dernburg et al. 1998). To maintain genome integrity, two events must occur: first, at least one DSB per chromosome pair must be repaired by interhomolog homologous recombination (HR) and resolved as a CO; second, additional DSBs must be repaired by HR with a non-CO outcome. Defects in either event promote genome instability, either from aneuploidy due to chromosome missegregation or through inappropriate DNA repair. Accordingly, numerous factors ensure the appropriate execution of meiotic HR, including those involved in the DDR (Lui and Colaiacovo 2013).
The covalent modification of the histone tails by a variety of posttranslational modifications (PTMs) confers exquisite variation in regulating DNA-dependent processes (Jenuwein and Allis 2001), including the DDR. Studies predominantly from mice and yeast have revealed that histone PTMs function at each step in the repair process. In response to exogenous DSBs, chromatin undergoes decondensation both locally and globally (Kruhlak et al. 2006; Dellaire et al. 2009). The relaxed chromatin structure facilitates the activation and recruitment of the kinase ATM, which initiates a signaling cascade leading to histone acetylation and additional chromatin remodeling. These modifications at the DSB site promote amplification of the DDR, recruit repair factors, and provide accessibility to the repair machinery (Deem et al. 2012). Following repair, acetylation of lysine 56 on histone H3 is required to inactivate the DDR (Chen and Tyler 2008) and to allow reassembly of nucleosomes at the repair site.
The interplay between chromatin structure, meiosis, and DNA repair remains a burgeoning area of research. In the Caenorhabditis elegans germ line, recent studies have linked global histone acetylation levels through cra-1 to meiotic DSB formation (Gao et al. 2015). However, the significance of specific histone acetylation marks to DSB formation and repair is not well understood. The acetylation of lysine 5 on histone H2A (H2AK5ac) is emerging as a vital player in these processes. In xnd-1 mutant germ lines, an increase in H2AK5ac was associated with decreased DSB formation, especially on the X chromosome (Wagner et al. 2010; Gao et al. 2015). H2AK5ac levels are diminished in htp-3 mutants and, in late pachytene nuclei, the recycling of H2AK5ac was correlated with local synaptonemal complex remodeling and repair of IR-induced lesions from sister chromatids (Couteau and Zetka 2011). These phenotypes of H2AK5ac are regulated, at least in part, by the histone acetyltransferase (HAT) mys-1, the worm homolog of Tip60, the catalytic subunit of the NuA4 HAT complex. Tip60 complexes are known to modulate the DDR through acetylation of the conserved sensor, ATM. ATM, in addition to its role in the DDR, has emerged as a component of a regulatory DSB feedback loop for meiotic DSBs, raising the possibility that Tip60, ATM, and H2AK5ac may share roles in both DSB formation and repair. We have found that xnd-1 provides a lens with which to explore the interplay between these factors in the C. elegans germ line.
Previously, we identified xnd-1 as an autosomally-associated protein that regulates X chromosome CO formation through chromatin structure (Wagner et al. 2010); additionally, we recently described a role for xnd-1 in germ line development (Mainpal et al. 2015). The variability in the severity of xnd-1 mutant phenotypes, including brood size, lethality, and sterility, was reminiscent of mutator loci and “mortal germ line” genes, and suggested a broader role for xnd-1 in maintaining genome stability. Here, we show that xnd-1 is a regulator of genome stability in the C. elegans germ line. xnd-1 mutants exhibited a mortal germ line phenotype and were sensitive to IR, consistent with a role in responding to DNA damage. Interestingly, a hypomorphic allele of the HAT mys-1 completely rescued xnd-1 IR sensitivity, improved fecundity, and restored meiotic DSB kinetics. This allele also reduced H2AK5ac levels, suggesting that these phenotypes may be a consequence of altered H2AK5ac levels. Although mys-1-dependent functions had no effect on the high incidence of male (Him) frequency of xnd-1 mutants, we found instead that the X chromosome CO defect in xnd-1 mutants was due to reduced expression of him-5, which XND-1 appears to regulate transcriptionally. This work suggests that XND-1 functions as both a modulator of transcription and chromatin structure, and identifies aspects of DSB formation and repair that are more acutely or less sensitive to perturbations in these functions.
Materials and Methods
Culture and strains
For all experiments, worms were cultured on NGM plates seeded with OP50 at 20° unless otherwise noted (Brenner 1974). Mutant strains used in this study were: LG I, hus-1(op244), atm-1(gk186), cep-1(gk138), cep-1(lg12501); LG III, xnd-1(ok709); LG IV, ced-3(n717); and LG V, mys-1(n3681), him-5(ok1896). Some strains were provided by the Caenorhabditis Genetics Center. xnd-1(ok709) was outcrossed multiple times for these studies due to long-term maintenance problems with the strain. Double and triple mutants were generated using standard genetic techniques and are listed in Supplemental Material, Table S1. Creation of transgenic animals is described below. N2 served as wild-type controls in this study. For strains containing either xnd-1(ok709) and/or an allele that must be balanced, F2 hermaphrodites were used unless otherwise noted. Due to some phenotypic differences between xnd-1/qC1 and xnd-1/hT2 populations, double and triple mutants were compared to isogenic balancer strains (xnd-1 F2 from hT2-balanced stock is described in Table 1; xnd-1 F2 from qC1-balanced stock is described in Table 2).
General characteristics of strains used in this study
| Genotype | N | Avg. Eggs ± SEM | Avg. Brood ± SEM | % Lethal ± SEM (Normalized) | % Male ± SEM | |
|---|---|---|---|---|---|---|
| A | N2 | 5 | 235.20 ± 12.72 | 231.00 ± 13.48 | 0.00 ± 1.04 | 0.10 ± 0.104 |
| B | xnd-1 | 23 | 103.13 ± 10.97 | 40.39 ± 8.06 | 67.71 ± 4.82 | 15.49 ± 2.65* |
| C | mys-1 | 5 | 255.60 ± 21.73 | 226.20 ± 20.17 | 9.81 ± 1.24 | 0.00 ± 0.00 |
| D | xnd-1;mys-1 | 32 | 151.53 ± 5.14** | 114.03 ± 4.76** | 23.57 ± 1.81** | 13.00 ± 1.66 |
| E | hus-1 | 8 | 230.75 ± 13.12 | 184.75 ± 12.46 | 18.63 ± 2.11 | 0.61 ± 0.19 |
| F | xnd-1;hus-1 | 21 | 110.10 ± 12.56 | 32.71 ± 5.87 | 73.67 ± 3.83 | 26.17 ± 4.53* |
| G | cep-1(gk138) | 10 | 192.30 ± 19.81 | 170.00 ± 19.88 | 10.93 ± 2.58 | 0.43 ± 0.34 |
| H | xnd-1;cep-1(gk138) | 26 | 102.15 ± 10.69 | 31.04 ± 4.85 | 70.77 ± 3.60 | 30.28 ± 3.41** |
| I | cep-1(lg12501) | 8 | 213.38 ± 13.10 | 191.13 ± 12.25 | 8.74 ± 1.18 | 1.04 ± 0.33 |
| J | xnd-1;cep-1(lg12501) | 22 | 119.50 ± 10.40 | 32.27 ± 5.88 | 75.65 ± 3.63 | 34.69 ± 5.65** |
| K | atm-1 | 5 | 250.60 ± 10.17 | 239.60 ± 8.78 | 2.50 ± 0.63 | 0.08 ± 0.08 |
| L | xnd-1;atm-1 | 21 | 135.86 ± 9.68* | 67.67 ± 9.79* | 51.88 ± 4.92* | 10.23 ± 1.69 |
| M | ced-3 | 8 | 257.38 ± 14.60 | 192.00 ± 14.30 | 24.47 ± 1.90 | 0.44 ± 0.21 |
| N | xnd-1;ced-3 | 26 | 94.04 ± 5.32 | 19.77 ± 2.74 | 79.64 ± 2.25* | 18.80 ± 2.48 |
| O | atm-1;mys-1 | 18 | 174.83 ± 8.56 | 162.33 ± 8.37 | 5.57 ± 0.87 | 0.13 ± 0.07 |
| P | xnd-1;atm-1;mys-1 | 23 | 119.26 ± 8.53 | 78.96 ± 7.39** | 33.94 ± 3.13** | 22.93 ± 2.27* |
| Genotype | N | Avg. Eggs ± SEM | Avg. Brood ± SEM | % Lethal ± SEM (Normalized) | % Male ± SEM | |
|---|---|---|---|---|---|---|
| A | N2 | 5 | 235.20 ± 12.72 | 231.00 ± 13.48 | 0.00 ± 1.04 | 0.10 ± 0.104 |
| B | xnd-1 | 23 | 103.13 ± 10.97 | 40.39 ± 8.06 | 67.71 ± 4.82 | 15.49 ± 2.65* |
| C | mys-1 | 5 | 255.60 ± 21.73 | 226.20 ± 20.17 | 9.81 ± 1.24 | 0.00 ± 0.00 |
| D | xnd-1;mys-1 | 32 | 151.53 ± 5.14** | 114.03 ± 4.76** | 23.57 ± 1.81** | 13.00 ± 1.66 |
| E | hus-1 | 8 | 230.75 ± 13.12 | 184.75 ± 12.46 | 18.63 ± 2.11 | 0.61 ± 0.19 |
| F | xnd-1;hus-1 | 21 | 110.10 ± 12.56 | 32.71 ± 5.87 | 73.67 ± 3.83 | 26.17 ± 4.53* |
| G | cep-1(gk138) | 10 | 192.30 ± 19.81 | 170.00 ± 19.88 | 10.93 ± 2.58 | 0.43 ± 0.34 |
| H | xnd-1;cep-1(gk138) | 26 | 102.15 ± 10.69 | 31.04 ± 4.85 | 70.77 ± 3.60 | 30.28 ± 3.41** |
| I | cep-1(lg12501) | 8 | 213.38 ± 13.10 | 191.13 ± 12.25 | 8.74 ± 1.18 | 1.04 ± 0.33 |
| J | xnd-1;cep-1(lg12501) | 22 | 119.50 ± 10.40 | 32.27 ± 5.88 | 75.65 ± 3.63 | 34.69 ± 5.65** |
| K | atm-1 | 5 | 250.60 ± 10.17 | 239.60 ± 8.78 | 2.50 ± 0.63 | 0.08 ± 0.08 |
| L | xnd-1;atm-1 | 21 | 135.86 ± 9.68* | 67.67 ± 9.79* | 51.88 ± 4.92* | 10.23 ± 1.69 |
| M | ced-3 | 8 | 257.38 ± 14.60 | 192.00 ± 14.30 | 24.47 ± 1.90 | 0.44 ± 0.21 |
| N | xnd-1;ced-3 | 26 | 94.04 ± 5.32 | 19.77 ± 2.74 | 79.64 ± 2.25* | 18.80 ± 2.48 |
| O | atm-1;mys-1 | 18 | 174.83 ± 8.56 | 162.33 ± 8.37 | 5.57 ± 0.87 | 0.13 ± 0.07 |
| P | xnd-1;atm-1;mys-1 | 23 | 119.26 ± 8.53 | 78.96 ± 7.39** | 33.94 ± 3.13** | 22.93 ± 2.27* |
Data were collected and analyzed as described in Materials and Methods. % lethality is normalized to N2. * P < 0.05 vs. xnd-1; ** P < 0.01 vs. xnd-1. Avg., average.
| Genotype | N | Avg. Eggs ± SEM | Avg. Brood ± SEM | % Lethal ± SEM (Normalized) | % Male ± SEM | |
|---|---|---|---|---|---|---|
| A | N2 | 5 | 235.20 ± 12.72 | 231.00 ± 13.48 | 0.00 ± 1.04 | 0.10 ± 0.104 |
| B | xnd-1 | 23 | 103.13 ± 10.97 | 40.39 ± 8.06 | 67.71 ± 4.82 | 15.49 ± 2.65* |
| C | mys-1 | 5 | 255.60 ± 21.73 | 226.20 ± 20.17 | 9.81 ± 1.24 | 0.00 ± 0.00 |
| D | xnd-1;mys-1 | 32 | 151.53 ± 5.14** | 114.03 ± 4.76** | 23.57 ± 1.81** | 13.00 ± 1.66 |
| E | hus-1 | 8 | 230.75 ± 13.12 | 184.75 ± 12.46 | 18.63 ± 2.11 | 0.61 ± 0.19 |
| F | xnd-1;hus-1 | 21 | 110.10 ± 12.56 | 32.71 ± 5.87 | 73.67 ± 3.83 | 26.17 ± 4.53* |
| G | cep-1(gk138) | 10 | 192.30 ± 19.81 | 170.00 ± 19.88 | 10.93 ± 2.58 | 0.43 ± 0.34 |
| H | xnd-1;cep-1(gk138) | 26 | 102.15 ± 10.69 | 31.04 ± 4.85 | 70.77 ± 3.60 | 30.28 ± 3.41** |
| I | cep-1(lg12501) | 8 | 213.38 ± 13.10 | 191.13 ± 12.25 | 8.74 ± 1.18 | 1.04 ± 0.33 |
| J | xnd-1;cep-1(lg12501) | 22 | 119.50 ± 10.40 | 32.27 ± 5.88 | 75.65 ± 3.63 | 34.69 ± 5.65** |
| K | atm-1 | 5 | 250.60 ± 10.17 | 239.60 ± 8.78 | 2.50 ± 0.63 | 0.08 ± 0.08 |
| L | xnd-1;atm-1 | 21 | 135.86 ± 9.68* | 67.67 ± 9.79* | 51.88 ± 4.92* | 10.23 ± 1.69 |
| M | ced-3 | 8 | 257.38 ± 14.60 | 192.00 ± 14.30 | 24.47 ± 1.90 | 0.44 ± 0.21 |
| N | xnd-1;ced-3 | 26 | 94.04 ± 5.32 | 19.77 ± 2.74 | 79.64 ± 2.25* | 18.80 ± 2.48 |
| O | atm-1;mys-1 | 18 | 174.83 ± 8.56 | 162.33 ± 8.37 | 5.57 ± 0.87 | 0.13 ± 0.07 |
| P | xnd-1;atm-1;mys-1 | 23 | 119.26 ± 8.53 | 78.96 ± 7.39** | 33.94 ± 3.13** | 22.93 ± 2.27* |
| Genotype | N | Avg. Eggs ± SEM | Avg. Brood ± SEM | % Lethal ± SEM (Normalized) | % Male ± SEM | |
|---|---|---|---|---|---|---|
| A | N2 | 5 | 235.20 ± 12.72 | 231.00 ± 13.48 | 0.00 ± 1.04 | 0.10 ± 0.104 |
| B | xnd-1 | 23 | 103.13 ± 10.97 | 40.39 ± 8.06 | 67.71 ± 4.82 | 15.49 ± 2.65* |
| C | mys-1 | 5 | 255.60 ± 21.73 | 226.20 ± 20.17 | 9.81 ± 1.24 | 0.00 ± 0.00 |
| D | xnd-1;mys-1 | 32 | 151.53 ± 5.14** | 114.03 ± 4.76** | 23.57 ± 1.81** | 13.00 ± 1.66 |
| E | hus-1 | 8 | 230.75 ± 13.12 | 184.75 ± 12.46 | 18.63 ± 2.11 | 0.61 ± 0.19 |
| F | xnd-1;hus-1 | 21 | 110.10 ± 12.56 | 32.71 ± 5.87 | 73.67 ± 3.83 | 26.17 ± 4.53* |
| G | cep-1(gk138) | 10 | 192.30 ± 19.81 | 170.00 ± 19.88 | 10.93 ± 2.58 | 0.43 ± 0.34 |
| H | xnd-1;cep-1(gk138) | 26 | 102.15 ± 10.69 | 31.04 ± 4.85 | 70.77 ± 3.60 | 30.28 ± 3.41** |
| I | cep-1(lg12501) | 8 | 213.38 ± 13.10 | 191.13 ± 12.25 | 8.74 ± 1.18 | 1.04 ± 0.33 |
| J | xnd-1;cep-1(lg12501) | 22 | 119.50 ± 10.40 | 32.27 ± 5.88 | 75.65 ± 3.63 | 34.69 ± 5.65** |
| K | atm-1 | 5 | 250.60 ± 10.17 | 239.60 ± 8.78 | 2.50 ± 0.63 | 0.08 ± 0.08 |
| L | xnd-1;atm-1 | 21 | 135.86 ± 9.68* | 67.67 ± 9.79* | 51.88 ± 4.92* | 10.23 ± 1.69 |
| M | ced-3 | 8 | 257.38 ± 14.60 | 192.00 ± 14.30 | 24.47 ± 1.90 | 0.44 ± 0.21 |
| N | xnd-1;ced-3 | 26 | 94.04 ± 5.32 | 19.77 ± 2.74 | 79.64 ± 2.25* | 18.80 ± 2.48 |
| O | atm-1;mys-1 | 18 | 174.83 ± 8.56 | 162.33 ± 8.37 | 5.57 ± 0.87 | 0.13 ± 0.07 |
| P | xnd-1;atm-1;mys-1 | 23 | 119.26 ± 8.53 | 78.96 ± 7.39** | 33.94 ± 3.13** | 22.93 ± 2.27* |
Data were collected and analyzed as described in Materials and Methods. % lethality is normalized to N2. * P < 0.05 vs. xnd-1; ** P < 0.01 vs. xnd-1. Avg., average.
eaIs15 [Ppie-1::him-5::gfp] rescues Him phenotype of xnd-1 mutants
| Genotype | N | Avg. Eggs ± SEM | Avg. Brood ± SEM | % Lethal ± SEM | % Male ± SEM | |
|---|---|---|---|---|---|---|
| A | eaIs15 | 6 | 228.17 ± 33.41 | 212.67 ± 30.96 | 4.69 ± 1.72 | 0.00 ± 0.00 |
| B | him-5 | 9 | 253.33 ± 15.20 | 170.00 ± 9.58 | 31.25 ± 1.91 | 31.87 ± 0.98 |
| C | him-5;eaIs15 | 8 | 247.75 ± 12.33 | 228.63 ± 11.55 | 5.88 ± 0.78* | 0.30 ± 0.17* |
| D | xnd-1 | 26 | 109.69 ± 4.97 | 55.00 ± 3.60 | 49.71 ± 2.24 | 17.26 ± 2.05 |
| E | xnd-1;eaIs15 | 24 | 87.79 ± 10.27 | 31.71 ± 5.15 | 71.14 ± 3.81 | 0.09 ± 0.08** |
| Genotype | N | Avg. Eggs ± SEM | Avg. Brood ± SEM | % Lethal ± SEM | % Male ± SEM | |
|---|---|---|---|---|---|---|
| A | eaIs15 | 6 | 228.17 ± 33.41 | 212.67 ± 30.96 | 4.69 ± 1.72 | 0.00 ± 0.00 |
| B | him-5 | 9 | 253.33 ± 15.20 | 170.00 ± 9.58 | 31.25 ± 1.91 | 31.87 ± 0.98 |
| C | him-5;eaIs15 | 8 | 247.75 ± 12.33 | 228.63 ± 11.55 | 5.88 ± 0.78* | 0.30 ± 0.17* |
| D | xnd-1 | 26 | 109.69 ± 4.97 | 55.00 ± 3.60 | 49.71 ± 2.24 | 17.26 ± 2.05 |
| E | xnd-1;eaIs15 | 24 | 87.79 ± 10.27 | 31.71 ± 5.15 | 71.14 ± 3.81 | 0.09 ± 0.08** |
Data were collected and analyzed as described in Materials and Methods. % lethality is normalized to N2 (Table 1, row A). * P < 0.01 vs. him-5; ** P <0.01 vs. xnd-1. Avg., average.
| Genotype | N | Avg. Eggs ± SEM | Avg. Brood ± SEM | % Lethal ± SEM | % Male ± SEM | |
|---|---|---|---|---|---|---|
| A | eaIs15 | 6 | 228.17 ± 33.41 | 212.67 ± 30.96 | 4.69 ± 1.72 | 0.00 ± 0.00 |
| B | him-5 | 9 | 253.33 ± 15.20 | 170.00 ± 9.58 | 31.25 ± 1.91 | 31.87 ± 0.98 |
| C | him-5;eaIs15 | 8 | 247.75 ± 12.33 | 228.63 ± 11.55 | 5.88 ± 0.78* | 0.30 ± 0.17* |
| D | xnd-1 | 26 | 109.69 ± 4.97 | 55.00 ± 3.60 | 49.71 ± 2.24 | 17.26 ± 2.05 |
| E | xnd-1;eaIs15 | 24 | 87.79 ± 10.27 | 31.71 ± 5.15 | 71.14 ± 3.81 | 0.09 ± 0.08** |
| Genotype | N | Avg. Eggs ± SEM | Avg. Brood ± SEM | % Lethal ± SEM | % Male ± SEM | |
|---|---|---|---|---|---|---|
| A | eaIs15 | 6 | 228.17 ± 33.41 | 212.67 ± 30.96 | 4.69 ± 1.72 | 0.00 ± 0.00 |
| B | him-5 | 9 | 253.33 ± 15.20 | 170.00 ± 9.58 | 31.25 ± 1.91 | 31.87 ± 0.98 |
| C | him-5;eaIs15 | 8 | 247.75 ± 12.33 | 228.63 ± 11.55 | 5.88 ± 0.78* | 0.30 ± 0.17* |
| D | xnd-1 | 26 | 109.69 ± 4.97 | 55.00 ± 3.60 | 49.71 ± 2.24 | 17.26 ± 2.05 |
| E | xnd-1;eaIs15 | 24 | 87.79 ± 10.27 | 31.71 ± 5.15 | 71.14 ± 3.81 | 0.09 ± 0.08** |
Data were collected and analyzed as described in Materials and Methods. % lethality is normalized to N2 (Table 1, row A). * P < 0.01 vs. him-5; ** P <0.01 vs. xnd-1. Avg., average.
Clutch size/brood size/lethality/him frequency
L4 hermaphrodites of a given genotype were individually plated and transferred to a clean plate every 12 hr until egg-laying ceased. After transfer, the number of eggs and L1s on the plate were counted and recorded. Each plate was then scored for the number of adult hermaphrodites and males 3–4 d later. Timepoint data from each individual parent was combined to give total eggs, total adult brood, and total males. Percent hatching was calculated by dividing total adults by total eggs and multiplying by 100. Percent lethality was then calculated by subtracting this value from 100. Percent lethality is normalized to N2 to account for ∼2% error in egg counts. To calculate percent male, the total number of males was divided by the total number of adults and multiplied by 100. The data are presented as the mean ± SEM from isogenic parents. Statistical tests used were the Student’s t-test or Mann–Whitney U test, depending on whether or not the data were normally distributed based on the results of the D’Agostino–Pearson normality test.
Sterility and mortal germ line analysis
For sterility assays, all progeny from several homozygous, hermaphrodite parents were plated individually. For strains containing either xnd-1(ok709) and/or another allele that must be balanced, F1 hermaphrodites (M+Z−) were selfed to generate homozygous (M−Z−) F2 progeny that were plated individually. Each plate was scored for the presence or absence of eggs and/or progeny 5 d postplating. Only plates in which the adult hermaphrodite was still present were included in the analysis. Hermaphrodites failing to lay a single egg were scored as sterile. Data from isogenic worms were combined to give the total numbers of sterile worms and hermaphrodites scored. To calculate percent sterility, the total number of sterile worms was divided by the total number of hermaphrodites and multiplied by 100.
The progeny of 10, independent F1 (M+Z−) animals were used to start 12 lines (M−Z−) that were passaged each generation by picking the first L4 animals to a new plate. If no progeny were present, the next generation was seeded by an immediate cousin (i.e., progeny of one of the 12 lines from the same F1 parent). This step is critical for assaying generational sterility: since a subset of F2 xnd-1 animals are sterile due to defects in PGC specification (Mainpal et al. 2015), random picking alone could lead to population loss. Population sizes were quantified every other generation by binning into size categories (sterile; 1–10 progeny; 10–20; 20–50; 50–100; and > 100). Populations were declared fully sterile when all 12 animals gave no progeny.
Microarray
Day 1 hermaphrodites were dissected in 1 × sperm salts (50 mM PIPES pH 7.0, 25 mM KCl, 1 mM MgSO4, 45 mM NaCl, and 2 mM CaCl2) with 0.5 mM levamisole. Fifty distal gonads from both N2 and xnd-1 were cleaved away from the maturing oocytes by cutting extruded gonads at the bend and collecting gonads in Trizol (Invitrogen) on ice. Samples were vortexed and frozen at −20° prior to RNA isolation and cDNA synthesis, which were performed as previously described (Fukushige and Krause 2012). Microarrays were performed at the Genomics Core of the National Institute of Diabetes and Digestive and Kidney Diseases using the C. elegans Genome Array (Affymetrix).
Gene expression analysis
Approximately 1000 (DNA repair genes and him-5) or 500 (eaIs4 and eaIs15 transgenes) day 1 adults of a given genotype were washed thrice in 1 × M9 buffer (3 g/L KH2PO4, 6 g/L Na2HPO4, 5 g/L NaCl, and 1 mM MgSO4), resuspended in Trizol (Invitrogen), and vortexed for ∼60 sec before being flash frozen and stored at −80°. Once all the samples were collected, the samples were thawed on ice, sonicated, and RNA was isolated by chloroform extraction and isopropanol precipitation. Samples were treated with DNase (Sigma #AMPD1) and reverse transcribed into cDNA (Protoscript m-MuLV First Strand cDNA Synthesis kit, NEB #E6300S) according to the manufacturer’s instructions. Quantitative real-time PCRs were performed on the Applied Bio Systems 7300 Real Time PCR System using SYBR Green chemistry (SensiMix SYBR Hi-ROX kit, Bioline #QT-605) with transcript-specific primers designed using GETPrime and Primer3 (Table S2; Koressaar and Remm 2007; Gubelmann et al. 2011; Untergasser et al. 2012). The reference genes rpl-32 and Y45F10D.4 (Hoogewijs et al. 2008) were used for normalization across samples and gene expression was analyzed using the ΔCT method (Livak and Schmittgen 2001). Results are presented as the average of combined data from three (DNA repair genes and him-5) or two (transgenes) independent biological replicates, which in turn are comprised of three technical replicates each.
IR sensitivity
L4 hermaphrodites were plated on each of four 6 cm plates at 30–100 worms/plate depending on genotype and IR dose. The following day, worms were exposed to 10, 50, or 100 Gy of IR from a 137Cs source (Gammacell1000 Elite, Nordion International Inc.). Twelve hours postirradiation, worms were individually plated and allowed to lay for 12 hr, at which point the number of eggs and L1s on the plate were counted. Each plate was scored for the number of adult progeny 3–4 d later. Survival was calculated as the total number of adult progeny divided by the total number of eggs/L1s relative to untreated worms ± SEM for a minimum of two independent experiments.
Immunofluorescence
Day 1 adult worms were dissected in 1 × sperm salts with 1 mM levamisole and fixed in 0.5% triton/1% PFA for 5 min in a humid chamber. Slides were then freeze-cracked and immersed in 100% ethanol for 1 min. Following fixation, slides were washed in PBST (1 × PBS with 0.1% Tween) and incubated in primary antibody (rabbit α-H2AK5ac, Cell Signaling #2576, 1:100; guinea-pig anti-SYP-1, 1:2000, gift from Anne Villeneuve; rabbit anti-RAD-51 1:50000, Novus Biologicals; or mAb414 mouse anti-nuclear pore complex, Abcam, 1:1000) overnight at 4°. The next day, slides were washed and incubated in secondary antibody (α-rabbit Alexa 568, 1:2000 or α-guinea pig Alexa 488, 1:2000) for at least 2 hr at room temperature in the dark. Slides were washed three times, in 1 × PBST, 1 × PBST with DAPI, and 1× PBST, and then mounted in Prolong Gold with DAPI (Life Technologies) and visualized by confocal microscopy (Nikon Instruments).
Images were analyzed using Volocity 3D software (PerkinElmer). Quantification of H2AK5ac levels was performed by determining the ratio of intensity between anti-H2AK5ac (FITC channel) and anti-nuclear pore staining (Texas Red channel) in a 59 × 59 pixel box proximal to the transition zone as shown in Figure 4. Values for each germ line were averaged for three independent squares to confirm uniformity of staining. Final ratios are the average of at least eight gonads per genotype.
Western blotting
For each genotype, a population of primarily adult worms was transferred from between 2 and 6 6 cm plates into a glass conical tube and washed thrice with 1 × M9. The remaining liquid was removed and the worm pellet was transferred to a 1.5 ml tube and flash frozen in liquid nitrogen. Pellets were thawed on ice, mixed with an equal volume of Laemmli Sample Buffer (Bio-Rad #161-0737) with 5% β-mercaptoethanol (Amresco M131), sonicated in a water bath for 2 min, heated at 95° for 10 min, then spun in a tabletop centrifuge for 5 min at maximum speed. Samples were resolved by 12% PAGE (TGX FastCast, Bio-Rad) and transferred to nitrocellulose in 20% methanol. The membrane was blocked in 5% nonfat milk/TBST (50 mM Tris-HCl pH 7.4, 150 mM NaCl, and 0.1% Tween-20) overnight at 4°. The next day, the membrane was washed in TBST and incubated in α-FLAG M2 (Sigma-Aldrich F1804, 1:5000 in 3% milk/TBST) for 2 hr at room temperature, followed by α-mouse HRP (1:50,000 in 3% milk/TBST) for 1 hr at room temperature. Products were visualized by enhanced chemiluminescence (ECL) (Invitrogen #WP20005) according to the manufacturer’s instructions. The membrane was then washed in mild stripping solution (200 mM glycine, 3.5 mM SDS, and 1% Tween-20, pH 2.2 adjusted with HCl) twice for 10 min each, followed by two washes in PBS for 10 min each and two in TBST for 5 min each, followed by blocking overnight as before. The membrane was incubated in α-E7 (tubulin, Developmental Studies Hybridoma Bank, 1:2000 in 0.2% milk/TBST) for 1.5 hr at room temperature, followed by α-mouse HRP (1:3000 in 0.2% milk/TBST) for 1 hr at room temperature. Products were visualized by ECL.
Transgene construction
The Ppie-1::him-5::gfp::pie-1 3′ UTR transgene (eaIs15) was constructed by subcloning the genomic region containing the him-5 open reading frame into pJK7 (John White lab) using 5′ SpeI and 3′ MluI restriction sites. The Phim-5::him-5::gfp transgene (eaIs4) was constructed by recombineering 2 × TY::GFP::3 × FLAG into the fosmid clone WRM0634bF01 at the C-terminus of the him-5 coding sequence (Mainpal et al. 2015). All transgenes were integrated into the unc-119(ed3) strain using microparticle bombardment (Praitis et al. 2001).
Data availability
The authors state that all data necessary for confirming the conclusions presented in the article are represented fully within the article.
Results
xnd-1 is required for maintaining genome stability
Initial studies identified xnd-1 for its role in CO distribution and meiotic chromosome segregation. Mutations in xnd-1 increased embryonic lethality (approximately half of the eggs failed to hatch) and increased the incidence of males (Him phenotype) compared to the wild type, evidence of X chromosome nondisjunction (Wagner et al. 2010). However, since we were concerned that xnd-1 affects genome stability, we outcrossed our lab stocks over 10 times and confirmed these phenotypes by following F2 hermaphrodites throughout their reproductive lifespan, taking note of the total number of eggs laid by each animal (clutch size) and the subsequent brood (total number of resultant adult progeny), including the number of males. Despite substantial variation in the severity of each phenotype among xnd-1 hermaphrodites, we observed an overall decrease in fecundity and fitness as measured by number of eggs laid compared to the wild type (Table 1, compare rows A and B) and hatching frequency, respectively. As expected, the number of males in xnd-1 broods was markedly increased, albeit to a lower frequency than previously reported (Wagner et al. 2010), which we attribute to outcrossing. Consistent with previous reports, a fraction of xnd-1 hermaphrodites were completely sterile and failed to lay a single egg (13.95%, P < 0.0001 vs.N2, Fisher’s exact test; Wagner et al. 2010; Mainpal et al. 2015).
The variability that is observed in xnd-1 mutants was reminiscent of mutations that cause a mortal germ line phenotype (Ahmed and Hodgkin 2000). Therefore, we set out to determine if the sterility and brood sizes associated with xnd-1 mutations become more severe upon passaging. Fecundity was assayed in 12 lines from each of 10 independent F1 xnd-1 animals (M+Z− from xnd-1/hT2) across 30 generations, or until populations were declared fully sterile (see Materials and Methods). As shown in Figure 1A, average brood sizes decreased and the incidence of sterility increased with progressive generations in xnd-1 mutants. After six generations, however, the brood size appeared to level off to an average size of ∼30 progeny per worm (Figure 1B). The percentage of sterile animals, however, continued to increase over 30 generations (Figure 1A). These differences may reflect different thresholds for xnd-1-dependent function in egg production vs. offspring viability. Collectively, these results clearly indicate that xnd-1 mutants exhibit a mortal germ line phenotype.
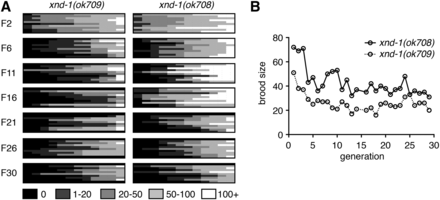
xnd-1 exhibits a mortal germ line phenotype. (A) Heat map depicts brood sizes of 12 lines (columns) from 10 independent xnd-1 F1s (rows) for indicated generations. Assay was performed as described in Materials and Methods. (B) Average brood sizes from fertile lines depicted in (A) over generations indicated. Both ok708 and ok709 alleles are presented and are phenotypically similar (Wagner et al. 2010). All other analyses are performed with ok709.
Close examination of brood dynamics also revealed periodic upswings and downswings in population size from single lines (Figure 1). For example, lines may have progressively decreased brood sizes over 5–10 generations, or have reached a size of fewer than 10 progeny and then increased to over 100 progeny in the next generation. Such transitions were reminiscent of our prior studies on rfs-1, a gene required for HR (Yanowitz 2008). Therefore, we set out to determine if xnd-1 may have a role in responding to DNA damage. To test this, we exposed xnd-1 F2 hermaphrodites to increasing doses of IR, which induces DSBs. The survival of progeny laid postexposure reflects the repair capacity in the hermaphrodite germ line. Survival of xnd-1 progeny post-IR was significantly decreased compared to wild-type at both 50 and 100 Gy (P < 0.01 vs.N2 at 50 and 100 Gy, Student’s t-test, Figure 2A). Collectively, these results suggest that xnd-1 is required for normal fertility, viability, and maintenance of genome stability.
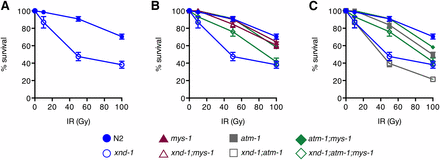
IR sensitivity of xnd-1, mys-1, and atm-1 mutants. Progeny survival 12–24 hr postirradiation in wild-type (N2, solid blue circles), xnd-1 (open blue circles), mys-1(n3681) (solid maroon triangles), xnd-1;mys-1(n3681) (open maroon triangles), atm-1(gk186) (solid gray squares), atm-1(gk186);xnd-1 (open gray squares), atm-1;mys-1 (solid green diamonds), and atm-1(gk186);xnd-1;mys-1(n3681) (open green diamonds). Data are plotted as the percent surviving progeny relative to untreated ± SEM (error bars). For easier viewing, the data are divided into N2 and xnd-1 (A), mys-1 and related strains in (B), and atm-1 and related strains in (C). IR, ionizing radiation.
We hypothesized that genome instability phenotypes in xnd-1 mutants may stem from a defect in DSB repair. First, we analyzed the quality of DSB repair by assessing DNA morphology in diakinesis-stage nuclei, as visualized by DAPI staining. In wild-type hermaphrodites, six condensed DAPI-staining bodies corresponding to six pairs of homologous chromosomes held together by chiasma are seen at diakinesis (Figure 3). Known DNA repair mutants, such as rad-51, exhibit decondensed chromatin and aggregates associated with defects in HR repair (Takanami et al. 2000; Rinaldo et al. 2002; Alpi et al. 2003). Interestingly, most xnd-1 diakinesis nuclei observed showed either the wild-type complement of six DAPI-staining bodies or seven DAPI-staining bodies consistent with nonexchange X chromosomes (∼62 and ∼17%, respectively; Figure 3A and Wagner et al. 2010). In most cases, chromatin appeared to be properly condensed. However, we observed chromatin abnormalities, including aggregation, decondensation, and fragments in ∼12% of nuclei (Figure 3A). We also observed nuclei with pachytene-like morphology clustered together at the −1 oocyte position, which we called “clustered nuclei” (∼10%; Figure 3 and Wagner et al. 2010). The frequency of abnormal oocytes and clustered nuclei were not sufficient to account for the embryonic lethality observed in xnd-1 mutants (Table 1, row B).
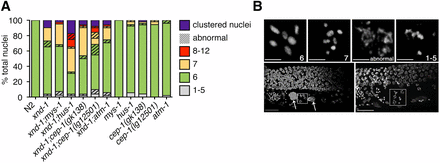
Cytological analysis and expression of select HR genes. (A) Quantification of the number of DAPI-staining bodies at diakinesis for indicated genotypes. Only the −1 oocyte was used for analysis (n = 40–52 nuclei). Color indicates the number of DAPI-staining bodies; hatched lines on top of a color indicate chromatin abnormalities. (B) Diakinesis phenotypes observed in oocytes of xnd-1 mutant animals. Shown are flattened images from a confocal Z-stack of the germ line region from DAPI-stained, whole-mount, xnd-1 mutant animals. Top row shows DNA morphologies in −1 oocytes. Scale bar, 2 µm. Bottom row shows the distal germ line on the top, proximal germ line below with sperm readily observed to the left. Large DAPI masses are marked with arrows (left); clusters of meiotic nuclei in the −2 (left) and −1 (right) oocyte positions are shown in the boxes. Normal diakinesis/diplotene nuclei can be seen in the −3 and −4 positions. Scale bar, 10 µm. DAPI, 4’,6-diamidino-2-phenylindole; HR, homologous recombination.
Because XND-1 protein is chromatin-associated (Wagner et al. 2010), we reasoned that XND-1 might alter the expression of DNA repair genes. We compared the transcriptional profile of xnd-1 mutant vs. wild-type germ lines using microarrays, focusing on genes encoding factors involved in HR-mediated DSB repair (reviewed in Lemmens and Tijsterman 2011). We found three DSB repair genes (rpa-2, gen-1, and slx-4/him-18) that were significantly downregulated in xnd-1 germ lines compared to wild-type (threshold of twofold, P < 0.05, Table S3). Consistent with their various roles in HR repair, all three factors exhibit sensitivity to DNA damaging agents (van Haaften et al. 2004; Saito et al. 2009; Bailly et al. 2010). We used quantitative PCRs (qPCRs) to verify the microarray data for the three DSB repair genes on our list, as well as other key genes functioning in the process such as rad-51, rad-54, and rtel-1 as controls (Livak and Schmittgen 2001; Gubelmann et al. 2011). In the germ lines of Day 1 adults, all the genes we assayed were expressed similarly between xnd-1 mutants and wild-type worms, except for rtel-1, whose expression was slightly reduced compared to wild-type (P = 0.037, Student’s t-test; Figure S1). As rtel-1 mutants display greater numbers of COs (Barber et al. 2008), but the overall number of COs in xnd-1 mutants is unchanged on the autosomes and reduced on the X (Wagner et al. 2010), it is unlikely that rtel-1 downregulation contributes to any xnd-1 phenotypes. Together, these results suggest that xnd-1 mutants show phenotypes consistent with genome instability, but these do not appear to stem from misregulation of canonical DSB repair genes.
A hypomorphic allele of mys-1 separates xnd-1 genome stability phenotypes from meiotic nondisjunction defects
The ability of xnd-1 mutants to form a CO on the X chromosome appears to be influenced by chromatin state (Wagner et al. 2010). Previous studies reported increased acetylation of lysine 5 on histone H2A (H2AK5ac) in xnd-1 mutant germ lines (Wagner et al. 2010; Gao et al. 2015). This accumulation of H2AK5ac is countered in part by RNAi against mys-1 (Wagner et al. 2010), suggesting that H2AK5 is an acetylation target of MYS-1, in agreement with in vitro data for Tip60 (Kimura and Horikoshi 1998; Wagner et al. 2010). Additionally, mys-1(RNAi) decreases the incidence of nonexchange X chromosomes at diakinesis in xnd-1 mutants (Wagner et al. 2010). To follow up on these studies and determine if mys-1 contributes to the genome instability phenotypes of xnd-1, we generated xnd-1;mys-1 double mutants. mys-1(n4075) is a presumptive null allele that is homozygous sterile with no diakinesis nuclei (Couteau and Zetka 2011). When combined with xnd-1, n4075 was epistatic, preventing the characterization of meiotic outcomes in the double mutant. Therefore, we used mys-1(n3681) (hereafter referred to as mys-1), which encodes a missense mutation (G341R) in the acetyltransferase domain and results in viable, homozygous progeny with at least a partially functional protein (Ceol and Horvitz 2004). Compared to xnd-1 alone, xnd-1;mys-1 hermaphrodites had significantly increased broods due to a combination of increased egg numbers and reduced lethality, although these phenotypes were not rescued to wild-type levels (Table 1, rows B and D). The incidence of sterility in F2 homozygotes was dramatically decreased in xnd-1;mys-1 mutants to < 1% (0.69% sterile, P < 0.0001 vs. xnd-1, Fisher’s exact test). These results indicate that mys-1 activity negatively impacts fecundity and fitness in xnd-1 mutants. Surprisingly, we did not observe a reduction in male frequency in xnd-1;mys-1(n3681) mutants as we expected, based on previous results with mys-1(RNAi) (Figure 3A and Table 1, 15.49% in xnd-1 vs. 13% in xnd-1;mys-1, P = 0.5688, Mann–Whitney) (Wagner et al. 2010), perhaps reflecting the weak loss-of-function nature of the mys-1(n3681) allele.
We next examined whether loss of mys-1 could suppress the IR sensitivity of xnd-1. We noted that mys-1(n3681) was not sensitive to IR (Figure 2B), which is in contrast to what is observed for Tip60 function in human cell lines, where the lack of Tip60 induced IR sensitivity (Kaidi and Jackson 2013). The lack of IR sensitivity of n3681 could be explained if the missense mutation only partially impairs acetyltransferase function or if other domains of MYS-1 are required for repair of IR-induced lesions. The lack of IR sensitivity of mys-1 allowed us to perform epistasis with xnd-1. mys-1(n3681) completely suppressed the IR sensitivity of xnd-1 mutant animals (Figure 2B), suggesting that mys-1 function contributes to the genome instability phenotypes of xnd-1.
As the n3681 allele encodes a missense mutation in the acetyltransferase domain of MYS-1, we hypothesized that the improved fitness we observed in xnd-1;mys-1 mutants could be due to decreased germ line H2AK5ac. We examined H2AK5ac in wild-type, xnd-1, mys-1, and xnd-1;mys-1 germ lines by immunofluorescence. Consistent with previous reports, we observed elevated H2AK5ac in the mitotic zone that decreased upon entry into meiosis in wild-type germ lines, yet remained elevated upon meiotic entry in xnd-1 germ lines (Figure 4 and Wagner et al. 2010; Gao et al. 2015). H2AK5ac was present in mys-1 germ lines, but its levels were diminished compared to wild-type (Figure 4). Quantification of H2AK5ac levels normalized to nuclear pore content showed a 30% reduction in mys-1 compared to wild-type. Consistent with these results, xnd-1;mys-1 mutant germ lines also showed a ∼30% reduction in H2AK5ac intensity in the pachytene region compared to xnd-1 alone (Figure 4), restoring H2AK5ac levels to close to wild-type. From these data, we infer that H2AK5 is an acetylation target of MYS-1 in the germ line and that n3681 hinders MYS-1 acetyltransferase activity. The presence of H2AK5ac in mitotic nuclei and developing oocytes of mys-1 mutants (data not shown) is consistent with prior analyses of mys-1(RNAi) and mys-1(n4075), and suggests that additional HATs acetylate H2AK5 in these regions (Wagner et al. 2010; Couteau and Zetka 2011). The suppression of xnd-1 germ line developmental defects and IR sensitivity, but not male frequency, raises the possibility that different thresholds of acetyltransferase activity exist for different MYS-1-dependent processes. Alternatively, these results could suggest that the HAT activity of MYS-1 is required to modulate xnd-1 IR sensitivity, but that additional function(s) of MYS-1, macromolecular complex formation for example, contribute to the meiotic nondisjunction phenotypes.
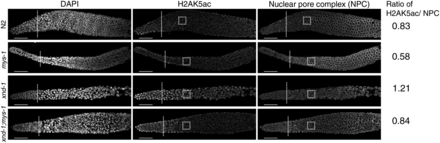
H2AK5 is an acetylation target of MYS-1. Immunofluorescence of DNA (left column), H2AK5ac (middle column), and nuclear pore complexes (right column, control) in wild-type (N2), mys-1, xnd-1, and xnd-1;mys-1 hermaphrodite germ lines. The distal end of the germ line is oriented left, and dotted lines mark meiotic entry based on transition zone morphology. All images were taken with identical camera and laser settings and processed identically. Quantification of H2AK5ac intensity was evaluated for the boxed regions and expressed as a ratio of nuclear pore staining (see Materials and Methods). Scale bar, 20 μm. DAPI, 4’,6-diamidino-2-phenylindole; IR, ionizing radiation; NPC, nuclear pore complex.
Fecundity and progeny survival in xnd-1 mutants partially depends on atm-1
Research has implicated Tip60 involvement in multiple levels of response to DNA damage, including signaling (Squatrito et al. 2006). Studies in mammalian cells have shown that, in response to either IR or treatment with Trichostatin A, which induces histone hyperacetylation through inhibition of class I and II histone deacetylases, Tip60 acetylates and promotes the activation of ATM kinase (Sun et al. 2005, 2009; Kaidi and Jackson 2013). We wondered if the reduced fitness of xnd-1 mutants could be explained by increased DNA damage signaling triggered by inappropriate accumulation of H2AK5ac. In the germ line, mitotic proliferation arrest and increased apoptosis are responses to genotoxic stress and governed by checkpoint genes, which ultimately prevent a cell with damaged DNA from continuing through the cell cycle until either the damage is repaired or apoptosis is initiated (Gartner et al. 2000). In C. elegans, both atm-1 and hus-1 are required for mitotic arrest following IR, possibly through parallel pathways (Hofmann et al. 2002; Garcia-Muse and Boulton 2005; Stergiou et al. 2007). Additionally, hus-1 is required for DNA damage-induced apoptosis through CEP-1-dependent transcriptional activation of egl-1 (Hofmann et al. 2002). Both atm-1 and hus-1 mutants exhibit genome instability phenotypes including a mortal germ line, high incidence of males, and spontaneous mutations (Hofmann et al. 2002; Jones et al. 2012), suggesting that they have roles in responding to endogenous genotoxic stress.
We employed double mutant analysis to examine whether DNA damaged-induced checkpoint pathways were hyperactive in xnd-1 hermaphrodite germ lines, leading to reduced fitness. Since cep-1 is required for DNA damage-induced apoptosis, but not mitotic arrest (Derry et al. 2001; Schumacher et al. 2001), we reasoned that we could distinguish the effects of mitotic arrest and DNA damage-induced apoptosis on xnd-1 fecundity through phenotypic differences between atm-1, hus-1, and cep-1 double mutants. Compared to xnd-1 alone, we observed no change in either the average clutch size of xnd-1;hus-1 and xnd-1;cep-1(gk138) hermaphrodites (Table 1, rows F and H) or the proportion of sterile hermaphrodites within these populations (15.79% sterile in xnd-1;hus-1 and 18.87% sterile in xnd-1;cep-1(gk138)). We observed similar results with a second allele of cep-1, lg12501, which also abrogates egl-1 induction, although retains some function lost by the gk138 allele (Table 1, row J, 8.33% sterile in xnd-1;cep-1(lg12501), P = 0.09 vs. xnd-1, Fisher’s exact test; Schumacher et al. 2005; Waters et al. 2010). These results suggest that neither hus-1-mediated DNA damage-induced mitotic arrest nor cep-1-mediated apoptosis contribute to the fecundity of xnd-1 hermaphrodites.
Interestingly, the frequency of male progeny in xnd-1;hus-1 and xnd-1;cep-1 populations was notably increased compared to xnd-1 (Table 1, rows B, F, H, and J), which could implicate a role for hus-1 and cep-1 in DSB formation. If this were the case, we would expect hus-1 and cep-1 to enhance the xnd-1 CO defect and increase the frequency of univalents in diakinesis oocytes. Compared to xnd-1, both xnd-1;hus-1 and xnd-1;cep-1(gk138) exhibited an increase in seven or more DAPI-staining bodies in the −1 oocyte (Figure 3A, P = 0.009 for xnd-1;hus-1 vs. xnd-1 and 0.027 for xnd-1;cep-1(gk138) vs. xnd-1, Z-test for proportions). Results for xnd-1;cep-1(lg12501) diakinesis oocytes were milder, with ∼6% of −1 oocytes exhibiting 8–12 DAPI-staining bodies, a phenotype never observed in xnd-1 single mutants (Figure 3A). The difference in diakinesis phenotypes between xnd-1;cep-1(gk138) and xnd-1;cep-1(lg12501) are opposite those reported for these cep-1 alleles and him-5 (Mateo et al. 2016), perhaps reflecting the more complex role of xnd-1 in DSB formation, influencing DSB timing, number, and distribution.
One explanation for the increase in males in xnd-1;hus-1 and xnd-1;cep-1 mutants is that DNA damage-induced apoptosis selectively eliminates nuclei that failed to receive a CO on the X chromosome. To test this, we examined physiological germ cell death, a second apoptotic pathway that culls ∼50% of germ line nuclei under normal conditions (Gumienny et al. 1999). Both physiological and DNA damage-induced cell death pathways rely on the core apoptotic machinery encoded by ced-3, ced-4, and ced-9 (Gartner et al. 2000). Therefore, we analyzed the male frequency of xnd-1;ced-3 double mutants to determine if apoptosis normally eliminates a subset of xnd-1 nuclei that fail to form X chromosome COs. We observed no change in male frequency between xnd-1 and xnd-1;ced-3 mutants (Table 1, rows B and N), ruling out that cell death selectively eliminates xnd-1 oocytes with nonexchange X chromosomes. Thus, these data support a role for hus-1 and cep-1 in DSB formation (Mateo et al. 2016).
In contrast to hus-1 and cep-1, atm-1 double mutants with xnd-1 exhibited both an increase in average clutch size compared to xnd-1 (Table 1, rows B and L, P = 0.03), and a significant reduction in the proportion of sterile animals (7.41% sterile, P = 0.0185 vs. xnd-1, Fisher’s exact test). xnd-1;atm-1 mutants also exhibited increased brood size and hatching compared to xnd-1, although male frequency remained unchanged (Table 1, rows B and L). Together, these results implicate a function of atm-1 as a factor contributing to fecundity and fitness in xnd-1 mutant germ lines.
mys-1 and atm-1 independently mediate xnd-1 phenotypes
Both xnd-1;mys-1 and xnd-1;atm-1 double mutants showed increased brood sizes and hatching rates compared to xnd-1 mutants, though the improvement was more pronounced in xnd-1;mys-1 double mutants (P < 0.0001 xnd-1;mys-1 vs. xnd-1;atm-1 for both phenotypes, Mann–Whitney, Table 1, rows B, D, and L). Given the function of Tip60 in ATM kinase activation in mammalian cells (Sun et al. 2005, 2009; Kaidi and Jackson 2013), we hypothesized that mys-1 and atm-1 may be functioning in the same pathway to control genome stability in xnd-1 germ lines. If this were the case, we would expect that atm-1, like mys-1, would suppress the IR sensitivity of xnd-1. Strikingly, however, IR sensitivity of xnd-1;atm-1 mutants resembled that of xnd-1 single mutants up to 50 Gy, suggesting that the cause of IR sensitivity in xnd-1 mutants is independent of atm-1 (Figure 2C). At 100 Gy IR, the sensitivity of xnd-1;atm-1 mutants was significantly lower than that of xnd-1 alone (P < 0.01, Student’s t-test), suggesting that atm-1 is required for survival following IR at high doses (> 50 Gy) only. In support of this, we noted that the IR sensitivity of atm-1 single mutants did not differ from that of wild-type worms until 100 Gy (P = 0.4804 at 10 Gy, P = 0.2715 at 50 Gy, and P = 0.0001 at 100 Gy).
The opposing IR sensitivities of xnd-1;mys-1 and xnd-1;atm-1 mutants provided an opportunity to examine an epistatic relationship between mys-1 and atm-1. We generated an xnd-1;atm-1;mys-1 triple mutant and assayed its sensitivity to IR (Figure 2, B and C). We observed that, up to 50 Gy IR, survival of xnd-1;atm-1;mys-1 mutants resembled that of xnd-1;mys-1 mutants, suggesting that the acetyltransferase domain of MYS-1 contributes to IR response independently of atm-1 and supporting the conclusion that atm-1 is not required for repair at low IR doses. By contrast, upon exposure to 100 Gy IR, survival of xnd-1;atm-1;mys-1 mutants was intermediate between the double mutants, with survival similar to xnd-1 mutants. These results are best explained by independent roles of mys-1 and atm-1 on xnd-1 survival postexposure to IR. These results also define a threshold IR dose that necessitates atm-1 function for survival, similar to what had previously been proposed for apoptosis induction (Stergiou et al. 2007).
In mammalian cells, a missense mutation in the chromodomain of Tip60 abolishes Tip60-dependent ATM activation in response to IR, yet retains housekeeping acetylation functions (Kaidi and Jackson 2013). Therefore, we wondered if mys-1 and atm-1 might function in additional pathways beyond those involved in survival following IR. To test this, we analyzed the clutch size, lethality, and male frequency of xnd-1;atm-1;mys-1 triple mutants (Table 1, row P). The average clutch size of xnd-1;atm-1;mys-1 triple mutants was significantly smaller than that of xnd-1;mys-1 mutants but similar to xnd-1;atm-1 mutants (Table 1, rows D, L, and P, P < 0.0029 vs. xnd-1;mys-1, P = 0.2308 vs. xnd-1;atm-1), suggesting that atm-1 is epistatic to mys-1 for this phenotype. However, we noticed a marked reduction in average clutch size between atm-1;mys-1 double mutants and either single mutant (Table 1, rows C, K, and O, P < 0.001 atm-1;mys-1 vs. atm-1 or mys-1), indicating that atm-1 and mys-1 function in parallel pathways to control egg production. Lethality of xnd-1;atm-1;mys-1 mutants fell between that of xnd-1;mys-1 and xnd-1;atm-1 mutants, and was statistically distinct from both (P < 0.01 xnd-1;atm-1;mys-1 vs. xnd-1;mys-1 or xnd-1;atm-1). Surprisingly, the male frequency of xnd-1;atm-1;mys-1 triple mutants was higher than that of either double mutant or each of the single mutants (Table 1, rows B, D, L, and P, P < 0.001 xnd-1;atm-1;mys-1 vs. xnd-1;mys-1 or xnd-1;atm-1, P < 0.05 xnd-1;atm-1;mys-1 vs. xnd-1). Consistent with the increase in male progeny, we observed an increase in the incidence of seven DAPI-staining bodies at diakinesis in xnd-1;atm-1;mys-1 germ lines (Figure 3A, P = 0.19 vs. xnd-1;mys-1, P < 0.01 vs. xnd-1;atm-1, Z test for proportions), implicating redundant roles for atm-1 and mys-1 in X chromosome CO formation. Taken together, these results are most consistent with a model in which atm-1 and mys-1 mediate genome stability phenotypes in xnd-1 mutants through independent mechanisms.
xnd-1 promotes X chromosome CO formation by regulating him-5 independently of mys-1
One proposed explanation for the X chromosome CO defect in xnd-1 mutants is that increased histone H2A lysine 5 acetylation changes the chromatin architecture such that the X chromosome is rendered inaccessible to DSBs. The previous observation that RNAi against mys-1 increases X chromosome CO formation (Wagner et al. 2010) supports this hypothesis, yet is confounded here by a similar male frequency (indicative of X chromosome nondisjunction) of xnd-1 and xnd-1;mys-1(n3681) broods (Table 1, rows B and D). This discord could reflect differences in mys-1 levels and activity between mys-1(RNAi) and mys-1(n3681); alternatively, it could reveal roles for a separate factor responsible for X chromosome CO formation.
A previous study suggested xnd-1 and him-5 function in the same genetic pathway in regards to X chromosome CO formation, and that HIM-5 levels and/or localization are dependent on xnd-1 (Meneely et al. 2012). To understand the nature of this regulation, we first used quantitative real-time PCR to assess the expression of him-5 in wild-type and xnd-1. We observed a dramatic reduction in him-5 transcript levels in xnd-1 mutants (Figure 5A, P < 0.001, Student’s t-test), raising the possibility that him-5 expression is regulated by XND-1 transcriptionally.
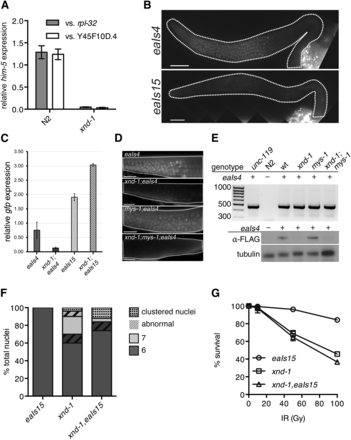
Downregulation of him-5 is responsible for the xnd-1 X chromosome CO defect. (A) qPCR analysis of him-5 expression in wt (N2) and xnd-1 hermaphrodites as compared to rpl-2 and y45f10d.4. (B) Germ line expression patterns of eaIs4 and eaIs15 recapitulate him-5 and pie-1, respectively. Images are taken with identical camera settings to directly compare expression levels. (C) qPCR analysis of eaIs4 and eaIs15 expression in wt and xnd-1 hermaphrodites as compared to y45f10d.4. (D) Germ line expression of eaIs4 is reduced in xnd-1. eaIs4 is visualized live by GFP fluorescence (green) in wt, xnd-1, mys-1, and xnd-1;mys-1 transgenic hermaphrodites. Scale bar, 10 μm. (E) xnd-1 is required for eaIs4 expression. Top: representative image of eaIs4 genotyping using transgene-specific primers shows the presence of eaIs4. Transgenic strains (indicated by +) show PCR product at ∼450 bp, while N2 controls do not. unc-119;eaIs4 is the founder strain, while wt is a wild-type control isolated from crossing eaIs4 into xnd-1;mys-1 mutants. Bottom: expression of eaIs4 visualized by western blot probed with α-FLAG antibody. Tubulin serves as a loading control. (F) Quantification of the number of DAPI-staining bodies at diakinesis for indicated genotypes. Only the −1 oocyte was used for analysis (n = 50 nuclei/genotype). Color indicates number of DAPI-staining bodies; hatched lines on top of a color indicate chromatin abnormalities. (G) Progeny survival 12–24 hr postirradiation in eaIs15 (wt, circles), xnd-1 (squares), and xnd-1;eaIs15 (triangles). The data are plotted as percent surviving progeny relative to untreated ± SEM (error bars). CO, crossover; DAPI, 4’,6-diamidino-2-phenylindole; GFP, green fluorescent protein; qPCR, quantitative polymerase chain reaction; wt, wild-type.
To test this directly, we compared the rescuing ability of two transgenes: the first, a him-5::gfp transgene driven by its native regulatory elements (Phim-5::him-5::gfp::3xFLAG, hereafter referred to as eaIs4), which was integrated as a fosmid into the genome; the second, a him-5::gfp fusion expressed from the pie-1 promoter (Ppie-1::him-5::gfp::pie-1 3′ UTR, hereafter referred to as eaIs15). Expression of eaIs4 recapitulates previously described HIM-5 localization patterns (Figure 5B and Meneely et al. 2012). Expression of eaIs15 recapitulates expression from pie-1 regulatory sequences (Merritt et al. 2008; Christopher Merritt 2010). HIM-5::GFP proteins from eaIs15 are diffusely localized in the nucleoplasm of mitotic zone nuclei, concentrate into puncta in the distal meiotic region, and again become diffusely nuclear near the gonad bend/diplotene (Figure S2). Diffuse nuclear staining from eaIs15 could also be seen in embryonic blastomeres, indicative of maternal deposition of HIM-5::GFP proteins/RNAs (data not shown). qPCR from whole worms showed a twofold higher abundance of eaIs15 transcripts in wild-type than in eaIs4 (Figure 5C), but live imaging showed more robust, early germ line expression of eaIs4 (Figure 5D). The disparity between RNA and protein levels in eaIs15 is likely due to late germ line accumulation of eaIs15 transcripts from the pie-1 promoter (Merritt et al. 2008; Christopher Merritt 2010). Despite differences in protein accumulation, both transgenes conferred rescue of the Him phenotype observed in him-5 mutants, suggesting they are fully functional (Table 2 and Table S4).
We next crossed the him-5 transgenes into the xnd-1 mutant background. Although PCR revealed the presence of eaIs4 in the genome (Figure 5E), qPCR showed a marked reduction in eaIs4 transcript level in the xnd-1 mutant background (Figure 5C). Consistently, both western blotting of whole animals (Figure 5E) and live imaging of GFP in adult germ lines (Figure 5C) failed to detect HIM-5::GFP proteins in the xnd-1 mutant, where these were readily observed in the wild type (Figure 5, D and E). Consistent with reduced expression, eaIs4 failed to suppress the Him phenotype conferred by xnd-1 (15.79% males in xnd-1 vs. 10.76% males in xnd-1;eaIs4, P = 0.07, Student’s t-test). By contrast, eaIs15 levels were not diminished in the xnd-1 mutants (Figure 5C) and HIM-5::GFP was robustly expressed in xnd-1 germ lines (Figure 5D). eaIs15 led to both a decrease in univalent X chromosomes (Figure 5F) and a reduction in the frequency of male progeny in xnd-1 mutants (Table 2). The difference in expression and concomitant rescue capability between him-5 expressed from its own vs. a heterologous promoter strongly argues that xnd-1 directly regulates him-5 transcriptionally. Collectively, these results indicate that a key role of xnd-1 in CO formation is in regulating him-5 expression.
Despite the rescue of the X chromosome CO defect, other xnd-1 phenotypes persisted in xnd-1,eaIs15 animals (Figure 5G and Table 2). The average clutch and brood sizes of xnd-1,eaIs15 mutants was indistinguishable from that of xnd-1 mutants (Table 2). We observed a small but significant decrease in hatching in xnd-1 mutants when eaIs15 was present, although it appears additive with the small increase in lethality observed in wild-type worms containing the transgene, suggesting that it may be due to the site of integration of the transgene. The presence of eaIs15 had no effect on the sensitivity of xnd-1 mutants to IR (Figure 5G). Together, these results reveal that decreased him-5 expression accounts for only the X chromosome CO defect observed in xnd-1 mutants. The failure of eaIs15 to rescue other xnd-1 phenotypes points to the involvement of multiple genes and/or pathways independent of him-5. Importantly, it also suggests that defects in meiotic DSB formation conferred by loss of him-5 are not sufficient to explain the lethality observed in xnd-1 mutants.
Having established that expression of him-5 is controlled by xnd-1, we wanted to test if mys-1 also affected him-5 expression. Therefore, we crossed eaIs4 into xnd-1;mys-1 mutants and probed for him-5 transgene expression. Similar to what we observed in xnd-1 mutants, expression of eaIs4 was barely detectable in xnd-1;mys-1 mutants (Figure 5, D and E). Consistent with diminished expression, eaIs4 was unable to rescue the Him phenotype of xnd-1;mys-1 (11.20% males in xnd-1;mys-1;eaIs4). These results indicate that him-5 expression is independent of mys-1-dependent changes in chromatin architecture; however, we cannot rule out the possibility that stronger reduction in mys-1 activity would elicit changes in him-5 expression.
xnd-1 control of DSB timing is mys-1-dependent
One of the most striking features of xnd-1 meiotic germ lines is a change in the timing of DSBs (Meneely et al. 2012; Gao et al. 2015), as monitored by accumulation of the HR strand-exchange protein RAD-51 (Mets and Meyer 2009). Unlike the wild type, in which a few RAD-51 foci can be seen in transition zone nuclei but in which significant RAD-51 accumulation is not observed until midpachytene (Figure 6A), in xnd-1, RAD-51 accumulates in transition zone nuclei reaching peak levels in very early pachytene (Figure 6A and Meneely et al. 2012). Prior studies showed that this pattern of RAD-51 accumulation was independent of global histone acetylation (Gao et al. 2015), but its relationship to specific changes in chromatin structure were not explored. We hypothesized that the change in DSB dynamics could be a reflection of the underlying chromatin structure in xnd-1 mutants and would therefore be suppressed by mys-1 mutation but not by ectopic him-5 expression (eaIs15), both of which alone displayed the wild-type pattern of RAD-51 accumulation (Figure 6, A and B). In support of this hypothesis, we observed that the premature accumulation of RAD-51 was suppressed in xnd-1;mys-1 double mutants (Figure 6A) but not xnd-1;eaIs15 (Figure 6B). These results support the model that DSB kinetics are shaped by changes in chromatin accessibility.
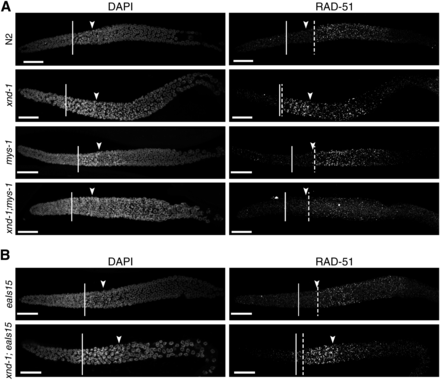
Altered dynamics of DSBs in xnd-1 are mys-1-dependent. Dissected germ lines were stained with DAPI to visualize DNA and anti-RAD-51 to visualize DSB repair intermediates. The beginning of the transition zone is marked with a solid line, the end with an arrowhead; onset of significant RAD-51 foci accumulation with a dashed line. (A) Kinetics of DSBs are altered in xnd-1 compared to wild-type, with high levels of RAD-51 appearing in the transition zone. DSB accumulation in mys-1 is similar to wild-type, peaking in midpachytene. mys-1 suppresses the formation of early DSBs in xnd-1. (B) Ectopic expression of him-5 cannot suppress the induction of early DSBs in xnd-1. RAD-51 accumulation appears normal in eaIs15 animals (top). xnd-1 is epistatic to eaIs15 and the double mutant shows accumulation of RAD-51 foci in the transition zone. Scale bar, 20 μm. DAPI, 4’,6-diamidino-2-phenylindole; DSB, double-strand break.
Discussion
xnd-1 is a model of genome instability
Here, we show that xnd-1, previously identified for its role in X chromosome DSB formation and germ line development (Wagner et al. 2010; Mainpal et al. 2015), is also an important regulator of genome stability in the C. elegans germ line. Homozygous xnd-1 hermaphrodites exhibit reduced fecundity in early generations that continues to decrease over time (Figure 1), a phenotype that is characteristic of factors involved in telomere maintenance, DNA damage sensing, and chromatin modification (Ahmed et al. 2001; Hofmann et al. 2002; Andersen and Horvitz 2007; Katz et al. 2009). The low broods of xnd-1 hermaphrodites result from a combination of decreased egg production and increased lethality (and perhaps reduced sperm count/function, although this has not been tested) that are independent of autosomal nondisjunction (Table 1 and Wagner et al. 2010). Consistent with a role in maintaining genome stability, xnd-1 mutants are sensitive to IR (Figure 2), suggesting that xnd-1 meiotic nuclei are unable to either properly respond to or repair exogenous DSBs. However, several observations suggest that xnd-1 mutants are competent for DSB repair. First, induction of exogenous DSBs by low dose IR restores X chromosome CO formation (Wagner et al. 2010), suggesting that the DSB repair machinery is functional. Second, the majority of diakinesis oocytes exhibit well-condensed chromosomes (Figure 3A), whereas repair mutants often fail to form distinct bivalents and show massive chromosome fusions and/or DNA fragments. Third, expression of HR genes appears unaltered (Figure S1 and Table S3). Together with the observations that a hypomorphic allele of mys-1 completely rescued xnd-1 IR sensitivity (Figure 2B and Table 1), these results argue that the genome instability phenotypes in xnd-1 cannot be explained by direct regulation of the HR machinery.
How does xnd-1 deficiency lead to genomic instability, if not through regulation of HR? Our data suggest that this may be through the alteration of germline chromatin.
A missense mutation that alters the acetyltransferase domain of MYS-1 significantly improved xnd-1 genome instability phenotypes. Our previous observation that mys-1(RNAi) reduced both germ line H2AK5ac and restored X chromosome CO formation in xnd-1 mutants suggested that mys-1 may exert its function through H2AK5ac (Wagner et al. 2010). The improved fecundity, fitness, and survival following IR in xnd-1;mys-1(n3681) mutants correlated with a reduction in H2AK5ac in meiotic nuclei, suggesting that the n3681 allele disrupts MYS-1 acetyltransferase activity and that this activity is required for the genome instability phenotypes of xnd-1.
We envision several possible models to explain the relationship between H2AK5ac, mys-1, and xnd-1 phenotypes (Figure 7). In the first model, XND-1 may negatively regulate mys-1 expression. In the absence of xnd-1, MYS-1 levels would be upregulated leading to IR sensitivity, altered DSB kinetics, and defects in DSB formation on the X chromosome (as suggested by Wagner et al. 2010). The hypomorphic mys-1 allele would reduce the increased germ line H2AK5ac levels to restore levels to near wild-type, sufficient to rescue all but the X chromosome CO defect. Regulation of cra-1 by xnd-1 could contribute to increased H2AK5ac levels (Lui and Colaiacovo 2013; Gao et al. 2015) in a mys-1-dependent fashion. cra-1 promotes global histone acetylation by antagonizing acetyl-CoA hydrolase ACER-1, leading to an increased pool of acetyl-CoA which MYS-1 (and other HATs) may then utilize to increase accumulation of H2AK5ac and other histone PTMs (Gao et al. 2015). This model predicts that different thresholds of mys-1 activity are required to potentiate different aspects of germ line function. Our second model proposes that xnd-1 regulates H2AK5ac levels independently of mys-1, perhaps through activation of a histone deacetylase. In the absence of xnd-1, deacetylase activity would be impaired and H2AK5ac would accumulate. Reduction of mys-1 function would counteract this accumulation by limiting the initial pool of H2AK5ac. In this model, different thresholds of H2AK5ac, or perhaps a balance of H2AK5ac and other acetylated histone marks, would potentiate different XND-1-dependent processes.
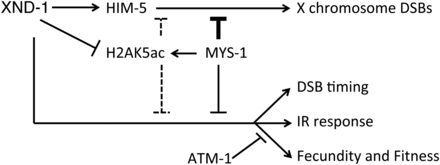
Model of genetic mediators of xnd-1 phenotypes. In addition to defects in X chromosome DSB formation (Wagner et al. 2010), xnd-1 mutants exhibit both changes in the timing of DSBs and genome instability phenotypes, including reduced fecundity and fitness and IR sensitivity. A partial loss-of-function allele of mys-1 improves these latter phenotypes but not the X chromosome DSB defect, in part through modulation of histone H2AK5 acetylation. Loss of atm-1 improves xnd-1 fecundity and fitness, and does not affect survival following IR until high doses, where it appears to be generally required. Our data suggest that mys-1 and atm-1 function in parallel pathways. The relationship between xnd-1, mys-1, and atm-1 is unknown. The reduced X chromosome CO formation in xnd-1 mutants is due to downregulation of him-5, which xnd-1 regulates transcriptionally. CO crossover; DSB, double-strand break; IR, ionizing radiation.
Finally, mys-1 may contribute to xnd-1 phenotypes through a mechanism apart from H2AK5 acetylation. An analysis of genetic interaction networks identified mys-1 as a “hub” gene, defined as a gene whose loss enhanced the phenotypic consequences of mutations in unrelated genes and functional pathways (Lehner et al. 2006). It is also possible that the difference in restoration of X chromosome CO formation in xnd-1;mys-1(RNAi) mutants but not in xnd-1;mys-1(n3681) mutants reflects the function of different MYS-1 complexes, only one of which functions as a HAT. Further studies are needed to elucidate the MYS-1 complexes and the mechanism by which mys-1 contributes to xnd-1 phenotypes.
Relationship of XND-1 and chromatin modifications to DNA repair
In late pachytene nuclei exposed to IR, H2AK5ac removal precedes localized desynapsis and correlates with intersister (IS) DSB repair, suggesting that turnover of this chromatin marks is necessary for the DDR (Couteau and Zetka 2011). By extension, we hypothesize that the decrease in H2AK5ac that we observe upon meiotic entry, a time that coincides with the onset of programmed DSB formation and HR repair, facilitates IS-HR for the excess DSBs that are made in meiotic nuclei. The elevated H2AK5ac levels in xnd-1 mutants could impede repair by preventing access to the sister chromatid and/or by interfering with checkpoint signaling. Recent data in yeast has suggested that IS repair precedes interhomolog repair (Joshi et al. 2015). Although it is unknown if this temporal distinction holds true for C. elegans, it is tempting to speculate that the increased RAD-51 accumulation at the onset of leptotene/zygotene in xnd-1 reflects the accumulation of DSBs destined for early IS repair events. The altered RAD-51 kinetics in xnd-1;mys-1 double mutants further supports such a model.
The checkpoint kinase ATM/atm-1 is also thought to function in IS repair (White et al. 2010; Couteau and Zetka 2011). Therefore, the increased lethality in response to IR may reflect redundant roles for these genes in IS repair. The improvements in fecundity and fitness that we observed in xnd-1;atm-1 counter the expected relationship based on IR sensitivity. These apposing responses to atm-1 knockdown support our supposition that the decreased brood sizes and embryonic lethality in xnd-1 do not result directly from defects in DNA damage repair, but rather from a checkpoint signaling function of an altered chromatin state. A key signal for DNA repair in mammals is γH2AX. This alternative histone is acetylated on lysine 5 to promote its removal and attenuate DNA damage signaling (Ikura et al. 2007). C. elegans lacks H2AX homologs. It is tempting to consider the possibility that H2AK5ac may serve an analogous signaling function.
Bipartate role for XND-1 in meiotic CO formation
Our results confirm that him-5 is sufficient to ensure DSB formation on the X in xnd-1 mutants (Figure 5). We found that him-5 is downregulated in xnd-1 mutants (Figure 5A), which explains why HIM-5 is undetectable in xnd-1 germ lines (Meneely et al. 2012). Furthermore, ectopic expression of him-5 under pie-1 regulatory elements is sufficient to restore X chromosome CO formation (Figure 5F and Table 2), indicating that XND-1 regulates him-5 transcriptionally.
The inability of the Phim-5::him-5::gfp transgene to suppress the Him phenotype of xnd-1 (Table 1) correlates with reduced expression of the transgene in the xnd-1 mutant background. The evidence for upregulation of him-5 and downregulation of cra-1 (Gao et al. 2013) in xnd-1 mutant animals suggests a role for XND-1 as a transcription factor, likely working with additional cofactors to positively and negatively regulate target genes. Future studies to identify XND-1 binding partners and their impact on him-5 and cra-1 transcriptional targets will likely provide important mechanistic insight into XND-1 function in a variety of germ line processes.
Despite an essential role for him-5 in promoting X chromosome COs, chromatin structure clearly still plays a critical role in establishing the recombination landscape. In wild-type germ lines, the X chromosomes receive fewer DSBs than autosomes (Gao et al. 2015), which may be attributed to the prevalence of repressive histone PTMs that facilitate transcriptional silencing (Kelly and Fire 1998; Kelly et al. 2002) limiting access to X chromosome DNA. Consistent with this model, mutations in the MES-2/3/6 complex that promotes H3K27 methylation on the X chromosome (Bender et al. 2004) desilence the X and suppress the Him phenotype of xnd-1 (Wagner et al. 2010). Despite evidence linking increased global acetylation to increased X chromosome DSB formation (Gao et al. 2013), xnd-1 mutants with increased H2AK5ac still receive dramatically fewer DSBs, both on the X chromosome and genome-wide. These data intimate that H2AK5ac hinders DSB formation. The function of this histone modification is largely unknown, but several reports implicate this mark in transcriptional activation (Shi et al. 2011; Rajagopal et al. 2014). One possibility is that H2AK5ac normally localizes to the active genes, which are found in preponderance in the central gene clusters on C. elegans autosomes (Barnes et al. 1995), facilitating their transcription but inhibiting COs. The increase in H2AK5ac levels in xnd-1 could cause this modification to spread to ectopic sites throughout the genome. A more uniform distribution of H2AK5ac could neutralize its effect on CO inhibition and drive COs to the most open regions of the genome, the autosomal gene clusters. Alternatively, however, H2AK5ac may promote DSB formation at specific sites in the genome, and the increased expression levels and ectopic localization of this modified histone could direct DSBs to these new sites. The suppression of the X chromosome CO defect by mys-1(RNAi) and not mys-1(n3681) hints that small changes in H2AK5ac can dramatically alter CO patterns. Despite not affecting CO formation on the X chromosome, mys-1(n3681) did alter the kinetics of DSB formation (and repair), suggesting that chromatin structure differentially effects CO distribution, timing, and frequency. Understanding the relationship between different chromatin modifications and these meiotic processes is an area of further investigation.
Acknowledgments
We thank Cynthia Wagner and Zebulin Kessler for their assistance in the construction and bombardment of the Ppie-1::him-5::gfp transgene, and Tetsu Fukushige and Weiping Chen for assistance with the microarrays. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health (NIH) Office of Research Infrastructure Programs (P40 OD010440). The anti-E7 tubulin antibody developed by M. McCutcheon and S. Carroll and was obtained from the Developmental Studies Hybridoma Bank, created by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the NIH and maintained at the Department of Biology The University of Iowa. Sequencing of transgenic constructs and the ced-3(n717) allele was performed in the Genomics Research Core at the University of Pittsburgh. Funding for these studies was provided by A New Scholar in Aging award from the Ellison Medical Foundation (AG-NS-0879-12) to A.G., State of Pennsylvania Formula Funds to J.L.Y., and an R01 from the National Institute of General Medical Sciences (R01GM104007) to J.L.Y.
Footnotes
Supplemental material is available online at www.g3journal.org/lookup/suppl/doi:10.1534/g3.116.035725/-/DC1.
Communicating editor: D. J. de Koning

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}